
Abstract
Non-small cell lung cancer (NSCLC) is one of the leading causes of cancer-related deaths worldwide. The genomic database for NSCLC is expanding rapidly, highlighting the importance of characterizing subpopulations from diverse regions. This study aims to identify and correlate genomic variants in patients with non-squamous NSCLC from two cities in Paraná, Brazil, and compare these findings with data from The Cancer Genome Atlas (TCGA) and the broader Brazilian population. We conducted a retrospective study sequencing tumor sample from 133 patients. An in silico analysis was performed for gene functional analysis. Additional data on tumor mutational burden (TMB), microsatellite status, and clinicopathological characteristics were also collected. The mutational load in the studied population was comparable to that of the TCGA cohort. However, gene expression profiles differed significantly, particularly for the EGFR, TP53, KRAS-NRAS, STK11-KEAP1, MTAP-CDKN2A/B, and PDL-1 genes. The gene expression profile in this study also showed marked differences from the general Brazilian population, with notably higher expression rates of EGFR and PDL-1. Specifically, considering the PDL-1 expression levels, 14% were classified as hyper-expressors, 33% as hypo-expressors, and 52% as non-expressors. These proportions were statistically distinct from global literature but aligned with the Brazilian profile. The genomic profile of patients with NSCLC in Paraná reveals a regional signature, characterized by a higher frequency of EGFR and TP53 mutations, along with elevated PDL-1 expression. These findings highlight the potential for regional variations in NSCLC, which could inform personalized treatment strategies for this population.
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide, posing a significant challenge for modern oncology. According to the Global Cancer Statistics for 2020, it accounted for 2.2 million new cases (11.4% of all cancer diagnoses), with 55% of these cases occurring in developing countries. Lung cancer also resulted in 1.8 million deaths annually, representing 18% of all cancer-related deaths (Lopes et al., 2015; Sung et al., 2021). The 5-year overall survival rate for metastatic non-small cell lung cancer (NSCLC) treated with conventional chemotherapy ranges from 7% to 10%, with median survival times typically around 9–10 months (Gerber and Schiller, 2013; Leighl, 2012; Schiller et al., 2002).
The mechanisms of carcinogenesis, tumor promotion, and progression are well-documented. The identification of signal transduction pathways associated with tyrosine kinase receptors, neoangiogenesis, immune responses, the tumor microenvironment, survival in distant tissues, antiapoptotic mechanisms, metastasis, and the ability of tumor cells to evade the immune system has expanded the clinical options for preventing tumor progression in patients with NSCLC (Hanahan and Weinberg, 2011).
Molecular tools are now capable of sequencing nearly all tumor DNA, enabling the identification of specific genetic defects through next-generation sequencing (NGS) (Lopes et al., 2015). This advancement has led to the development of targeted molecular therapies, resulting in significant benefits for various oncological outcomes (Camidge et al., 2021; Drilon et al., 2020; Mok et al., 2009; Paik et al., 2020; Peters et al., 2017; Planchard et al., 2017; Rosell et al., 2012; Shaw et al., 2020, 2014; Skoulidis et al., 2021; Soria et al., 2018; Wolf et al., 2020). Building on this knowledge, several mechanisms involving immune response components have been identified in patients with NSCLC. These include defects in antigen presentation, selection of tumor clones with high mutational burdens, epigenetic silencing, hypermethylation that promotes neoantigens, alterations in oncogenic drivers, upregulation of genes that enhance immunosuppression in the tumor microenvironment, and the exhaustion of essential immune effector cells. Together, these factors contribute to a reduction in tumor-infiltrating lymphocytes in lung cancer (Anichini et al., 2020; Chen et al., 2022; McGranahan et al., 2017).
As these mechanisms can impede effective antitumor cellular responses, particular emphasis has been placed on the inhibition of T lymphocyte function by immune checkpoints such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed death 1 (PD-1). The binding of ligands to these co-receptors disrupts antigen recognition, which is essential for the cytotoxic activity of lymphocytes against tumors (Freeman et al., 2000; Keir et al., 2008). Consequently, their overexpression in neoplastic cells inhibits immune cell infiltration and functionality, creating immunological deserts that promote tumor progression (Dong et al., 2002; Vesely et al., 2011).
Given the continuous expansion of the global genomic database on lung cancer, it is essential to further investigate the profiles of specific populations. This study aims to describe and analyze the correlation between genomic variants and clinicopathological markers in patients with non-squamous NSCLC from a Brazilian population. Specifically, it will focus on EGFR mutations and PDL-1 expression, comparing these findings to the global genomic profile from The Cancer Genome Atlas (TCGA) dataset and exploring their clinicopathological significance.
Methods
This is a retrospective, observational, longitudinal analysis conducted on patients treated at three cancer centers in two cities in Paraná state, Brazil (Londrina and Curitiba), serving a combined population of over 2 million inhabitants. Data were collected from medical records over an 11-month period, from June 2022 to April 2023. The study proposal was approved by the Institutional Ethics and Research Committee (CAAE number 22943019.1.0000.0107, State University of Western Paraná). Informed consent was obtained from living patients or their legal guardians for deceased patients, ensuring that all participants were provided with information regarding confidentiality and data protection.
The study consecutively recruited 103 patients from the ProOnco—Oncoclínicas Londrina clinic, 20 from the CIONC clinic (Curitiba, Paraná), and 10 from the Hospital de Clínicas of the Federal University of Paraná (Curitiba, Paraná). Molecular information for the patients was obtained directly from genomic test reports, with no additional curation performed based on the findings. Data from medical records were systematically collected, including the following parameters: age at diagnosis, gender, smoking status, tobacco load, body mass index, histological subtype from biopsy, and disease stage.
Inclusion criteria for the study included patients over 18 years of age with an anatomopathological and immunohistochemical (IHC) diagnosis of non-squamous NSCLC; somatic molecular assessment of the tumor; locoregional disease amenable to potentially curative treatment; or metastatic disease measurable by imaging according to the Response Evaluation Criteria in Solid Tumors version 1.1. Additionally, patients must not have received prior systemic therapy. To assess PD-L1 status, we used the tumor proportion score (TPS), an internationally recognized method that standardizes the interpretation of the different antibody clones used (22C3, 28-8, and SP263), enabling consistent comparison across assays.
An in silico study was conducted using FunRich 3.1 software for functional analysis. The TCGA database was utilized to obtain the global gene mutation profile, while the TIMER 2.0 tool (available at http://timer.cistrome.org/) was used for comparative analysis with the sample, focusing on the gene mutation module. Two-way analysis of variance was applied for statistical analyses to compare gene profiles and mutational loads between samples. The comparative frequency of genes was analyzed to identify upregulated and downregulated genes, using a fold change >2 and the Mann–Whitney U test. The correlation profile among genes was assessed using Spearman’s correlation.
The sample was characterized using descriptive statistics, and the chi-square test was applied to assess significant statistical differences among the categories of each variable. Chi-square tests of independence were conducted to evaluate the association between the initial stage of the disease (locoregional or metastatic) and treatment type (adjuvant versus first-line treatment). Fisher’s exact test was used to determine the significance of each variable, both positive and negative.
For the correlation analysis, genes with mutations in more than 15 individuals were selected and underwent correspondence analysis to evaluate their potential influence on molecular diagnoses. Due to a substantial number of patients with indeterminate or inconclusive results, particularly concerning tumor mutational burden (TMB) and microsatellite instability (MSI), a refinement of the data was performed to enhance the relevance of the study findings. This involved a statistical reanalysis that excluded these data and recalculated the P values. Statistical analyses were conducted using XLSTAT 2017.19.02 and GraphPad Prism 9.0, with a significance level set at P < 0.05.
Results
A total of 133 patients were analyzed (see Table 1). The majority of participants were aged between 60 and 69 years (30%) and 70 and 79 years (31.5%) (P < 0.0001). Additionally, 57% of the participants were female (P = 0.083). Among the patients, 52.9% were smokers, and 46% reported a smoking history of more than 10 years per pack (P = 0.513). The most common histological subtype was adenocarcinoma, accounting for 98% of the sample (P < 0.001).
Clinicopathological Characteristics of Patients
N, number of individuals; %, percent of individuals.
Of the patients, 90 (74.3%) had metastatic disease, while 31 (25.6%) had localized or locoregional disease. Association tests regarding the initial stage of the disease revealed no significant differences. Molecular assessments using NGS—Foundation One CDx were performed for 78 of the 133 patients, representing 59% of the total exams in the sample. Additionally, 76 mutated genes, considered infrequent in this cohort (each with an incidence of <5% in the reports), were identified, typically found in the examinations of a single patient (see Table 2).
Genomic Variants Identified in the Study Population
N, number of individuals; %, percent of individuals.
The comparative gene profile of the study population versus that of the TCGA is shown in Figure 1A. Approximately 33% of patients exhibited PDL-1 hypo-expression (Fig. 1B). The distribution of genomic variants for each gene is depicted in Figure 1C, with EGFR mutations accounting for 33% of the total. Principal components correspondence analysis revealed 2 distinct subpopulations (Fig. 1D). The first subpopulation, located in quadrants 2 and 3, suggests that the predominant gene signature includes patients with mutations in EGFR and TP53 but not in KRAS or NRAS. Conversely, the analysis indicated that when mutations in KRAS or NRAS were present, EGFR and TP53 mutations were absent. The second subpopulation, in quadrants 1 and 4, confirmed that individuals with mutations in the MTAP gene frequently also carried co-mutations in CDKN2A/B, a phenomenon observed in 100% of the study participants.
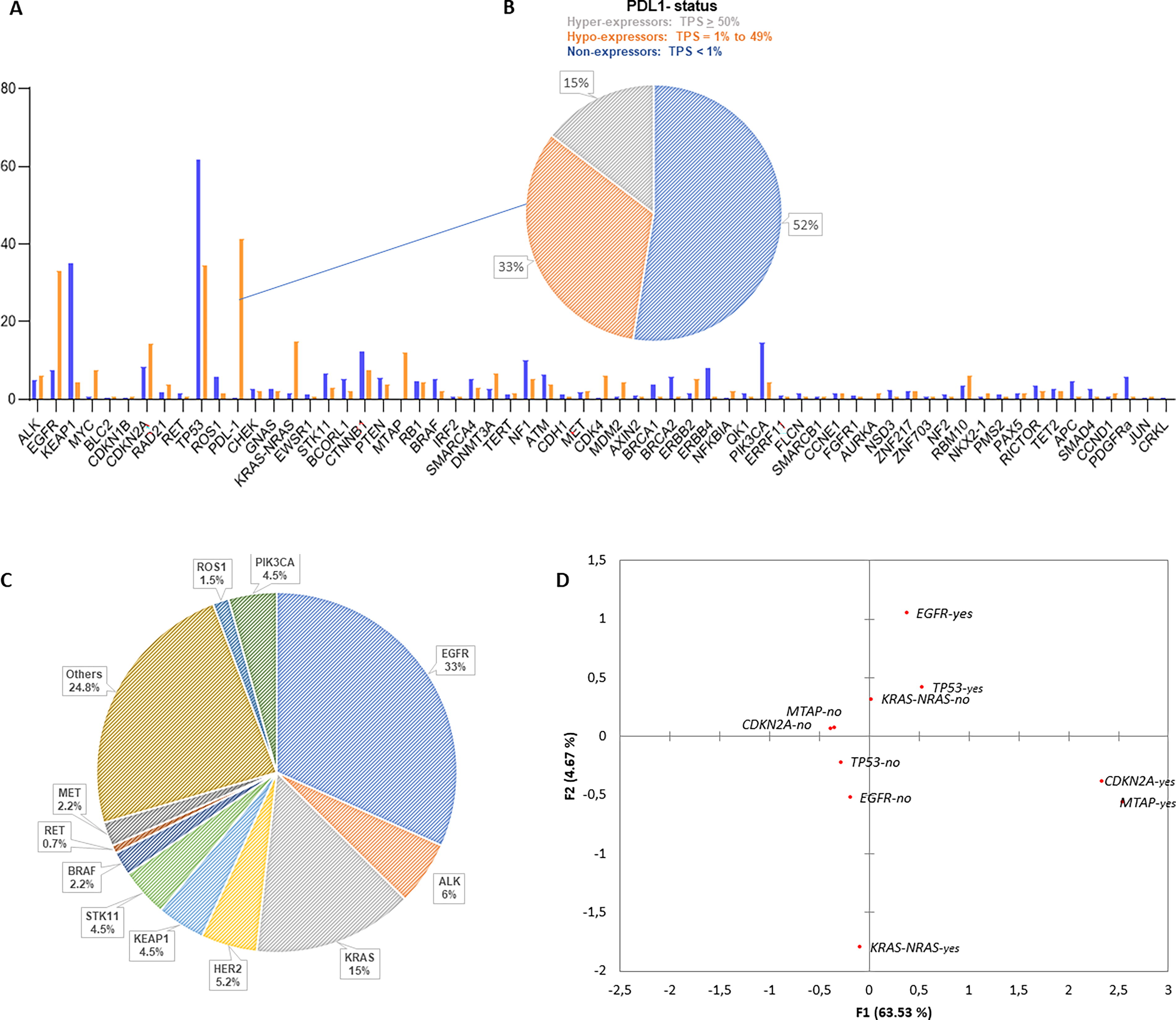
Gene profile overview.
In the in silico functional analysis, a comparative evaluation of the mutational profiles of the sequenced genes in the study sample and the TCGA was conducted (Fig. 2). The analysis revealed that the mutational load between the two populations did not differ significantly (P > 0.05). However, the expression profile of the mutated genes showed a significant difference (P < 0.001). The biological processes and signaling pathways associated with the genes identified in the study sample are illustrated in Figure 2A and B. Their cellular functions include cell communication (43.6%) and signal transduction (43.6%), with a focus on signaling pathways such as interferon-gamma (IFN-γ), hepatocyte growth factor receptor (c-Met), platelet-derived growth factor (PDGF), insulin-like growth factor 1 (IGF-1), erythroblastic leukemia viral oncogene homolog (ErbB), and membrane-associated estrogen receptor signaling.

Biological processes
Additionally, significant correlations were observed among mutated genes with a fold change >2, particularly between BRCA1 and SMAD4, AURKA and NSD3, MDM2 and CDK4, and BCORL1 and STK11. Notably, no association was found between EGFR and KRAS-NRAS (Fig. 3).
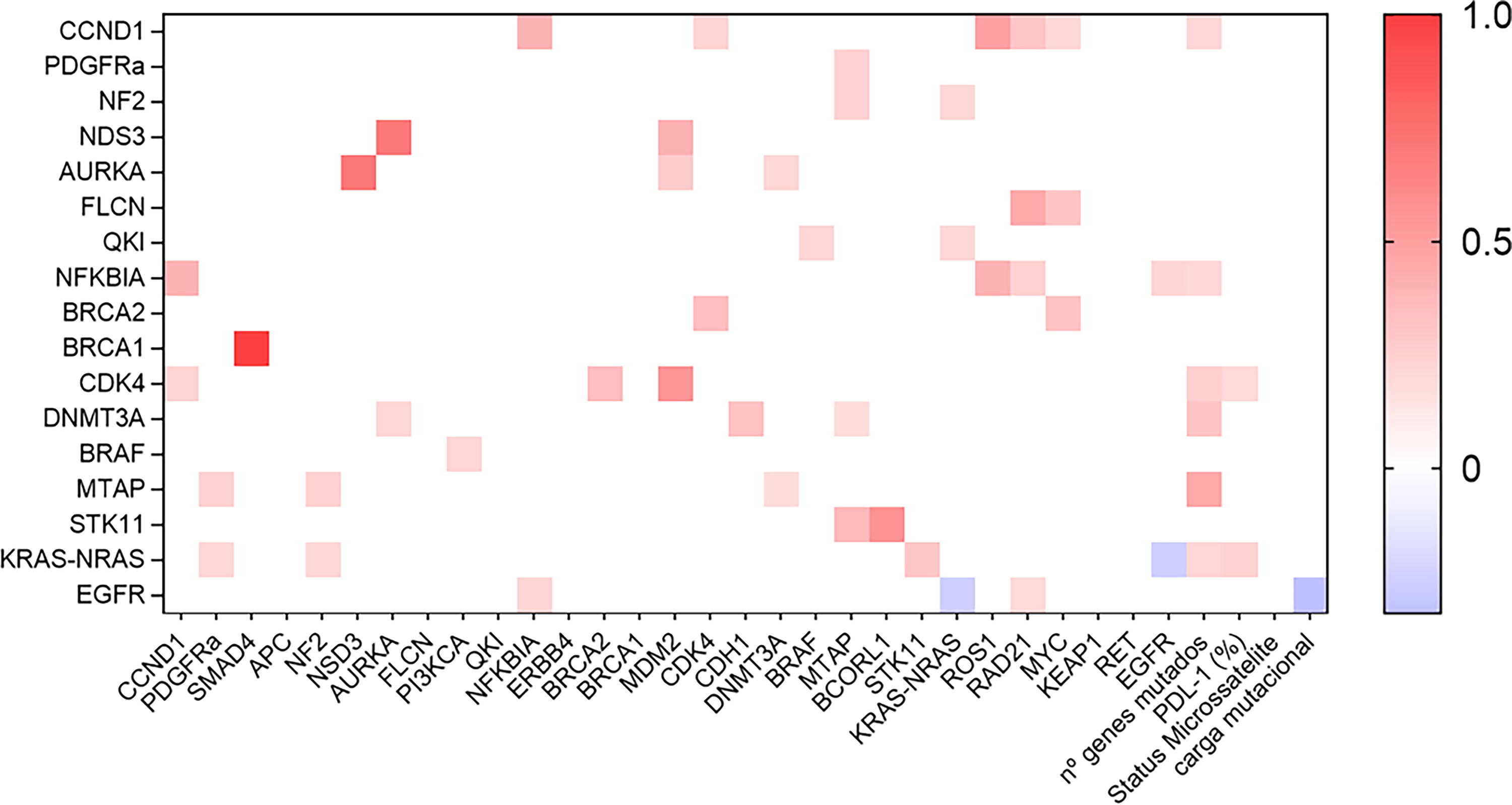
Profile of significant correlations (P < 0.05) between the genes sequenced in the patient sample. Heatmap built in GraphPad Prism 9.0 software based on correlation values (R) obtained through Spearman correlation between the genes expressed in the sample, the number of mutated genes, the expression of PDL-1 by immunohistochemistry, and the mutational load. The blue rectangles represent negative correlation, and the red ones represent positive correlation. The stronger the correlation, the more intense the red color of the rectangle; the weaker it is, the bluer the color.
The comparative analysis of gene frequency between the patient sample and TCGA samples is presented in Table 3, highlighting PDL-1 expression as the most significant, with a fold change of 105.2. Additionally, a comparison was conducted between the incidence of genomic variants identified in the samples and the data available in both global and Brazilian data (see Tables 4 and 5) (Awaya et al., 2004; Camidge et al., 2021; Chen et al., 2022; Paik et al., 2020; Planchard et al., 2017; Shaw et al., 2014; Wolf et al., 2020; Zhao et al., 2017). A total of 59 individuals had 1–3 mutated genes (44.36% of the sample), and 34 individuals had 4–6 mutated genes (25.56% of the sample, P < 0.001).
Comparative Frequency of Genes Sequenced in the Patient Sample and in The Cancer Genome Atlas Samples
Upregulated genes are represented by positive values, while downregulated genes are indicated by negative values.
TCGA, The Cancer Genome Atlas; %, percent.
Comparative Analysis of the Identified Genomic Variants Between the Study Population and The Cancer Genome Atlas Data
Gene expression differed significantly between the study population and the TCGA dataset (P = 0.0134).
Comparative Analysis of the Identified Genomic Variants Between the Study Population and Brazilian Population Data (Mascarenhas et al., 2021)
Gene expression differed significantly between the study population and the TCGA dataset (P = 0.0195).
Ninety individuals were diagnosed with metastatic disease (74.38%) (P < 0.001). Of these, 25 patients received adjuvant treatment (20.66%), while 97 patients underwent first-line systemic treatment (80.83%) (see Table 6). Regarding biomarker response (BMR), 87.3% of individuals exhibited low BMR, and 12.7% demonstrated high BMR (P < 0.001). Notably, no patients presented with microsatellite instability (MSI).
Mutational Status, Microsatellite Status, Programmed Death-Ligand 1 Expression, and Number of Tumor Mutations in the Study Population
N, number of individuals; %, percent of individuals; TMB, tumor mutational burden.
To explore the potential connection between TMB and smoking, as well as the inversely proportional relationship between TMB and the presence of clinically relevant driver mutations in NSCLC, a correlation analysis was conducted. A significant correlation was found between TMB and driver mutations, as well as between smoking and driver mutations, indicating an opposing relationship: When a driver mutation was identified, it was not associated with high BMR or a smoking habit. However, no correlation was detected between BMR and smoking (see Table 7). Regarding the PDL-1 variable (Table 8), 17 individuals were identified as hyper-expressors (14.6%), 38 as hypo-expressors (32.7%), and 61 as non-expressors (52.5%) (P < 0.0001). A comparison of the relative frequencies of PDL-1 expression in our sample with data from global and Brazilian data indicated that our patient sample exhibited higher PDL-1 expression (P = 0.040, Table 8).
Spearman Correlation Between Tumor Mutational Burden Variables, Smoking, and Presence of Driver Mutations in Non-Small Cell Lung Cancer in the Study Population
TMB, tumor mutational burden.
Comparative Analysis of PDL-1 Expression Between the Study Population and the Memorial Sloan Kettering Cancer Center and EXPRESS Studies (Schoenfeld et al., 2020; Dietel et al., 2019)
MSKCC, Memorial Sloan Kettering Cancer Center.
Discussion
The primary objective of this research was to elucidate the genomic landscape of patients with NSCLC from a specific segment of the population in two cities in Paraná, Brazil. Our study demonstrated that the mutational profile of the sample differs from both the currently cataloged genomic data and that of the TCGA. However, the frequency of gene variants shows some similarities with both the Brazilian and global populations. Notably, the majority of TCGA lung carcinoma samples are derived from surgical specimens, with 81% of the patients in this cohort being either active smokers or ex-smokers (The Cancer Genome Atlas Research Network, 2014).
In our sample, we identified a high prevalence of mutations in TP53 and EGFR, occurring in 59% and 33% of patients, respectively. In contrast, the prevalence of mutations in KRAS, STK11, and KEAP1 was low at 15%, 4.5%, and 4.5%, respectively. These findings highlight a distinct regional mutation profile within the studied population (Jordan et al., 2017; The Cancer Genome Atlas Research Network, 2014). The EGFR mutation was identified in 33% of cases, which is statistically different from the prevalence reported in worldwide and Brazilian studies (Jordan et al., 2017; Mascarenhas et al., 2021). These findings indicate that this population is primarily composed of smokers but also exhibits a high incidence of this activating mutation.
In Brazilian studies, Freitas and others reported EGFR mutations in 25.4% of 1,316 sequenced lung adenocarcinomas, while Cronemberger and others found EGFR alterations in 24% of 391 individuals within the same year. Additionally, Andreis et al. identified EGFR mutations in 19.2% of 619 patients with adenocarcinoma in a southern multicenter study. Similarly, Mascarenhas et al., using data from the Brazilian Thoracic Oncology Group (GBOT), confirmed a comparable prevalence of this mutation at 22.5% among 513 sequenced patients (Andreis et al., 2019; Cronemberger et al., 2020; Freitas et al., 2020; Mascarenhas et al., 2021).
Following the diagramming of genes and their interconnections within the interactome, as well as the principal component correspondence analysis, a distinct regional genomic signature emerged. A driver mutation in EGFR was found to pair with a co-mutation in TP53, which was mutually exclusive with mutations in KRAS and NRAS. This negative association has been observed in several retrospective studies; for example, research from Memorial Sloan Kettering Cancer Center indicated that only 2 out of 860 patients sequenced exhibited concurrent mutations in both EGFR and KRAS (Chen et al., 2022; Jordan et al., 2017; Mascarenhas et al., 2021).
The association between EGFR and TP53 mutations is well established, with TP53 frequently serving as a primary partner in genomic evaluations of populations enriched for EGFR mutations. This early truncal co-mutation, characterized by loss of function due to homozygosity or heterozygosity, is diagnosed in 54.6%–64% of metastatic NSCLC cases. It is associated with shorter disease-free intervals and therapeutic resistance, leading to a reduced response time to anti-EGFR tyrosine kinase inhibitors (TKIs). Vieira and others reported that mutations in EGFR were present in 57.1% of patients with tumors harboring somatic mutations in TP53. Additionally, other co-mutations in the EGFR landscape have been identified, including RB1 (9.5%–12.5%), CTNNB1 (5.3%–9.6%), PIK3CA (9%–12.4%), NKX2-1 (12.2%–16.7%), as well as CDK4, CDK6, and CCNE1 (Mascarenhas et al., 2021; Skoulidis and Heymach, 2019; Vieira et al., 2021).
TP53, a tumor suppressor gene, is mutated in ∼50% of NSCLC cases. This mutation is often associated with increased lymphatic dissemination and poorer prognosis in metastatic disease (Mogi and Kuwano, 2011; Schoenfeld et al., 2020). In our sample, this mutation was identified in 34% of individuals, representing 59% of the patients who underwent genomic evaluation using Foundation One CDx. This finding is consistent with previously reported literature. Recent advancements in the understanding and treatment of NSCLC have underscored the importance of PD-L1 expression and mutations in the EGFR and TP53 genes. These biomarkers not only influence tumor behavior but also inform therapeutic strategies. A retrospective analysis of 273 patients with NSCLC revealed that 24.9% exhibited positive PD-L1 expression. Notably, EGFR mutations were identified in 63% of cases and showed an inverse association with PD-L1 expression. This suggests that EGFR-mutant tumors may possess distinct immune evasion mechanisms, potentially affecting their responses to immunotherapy (Liu et al., 2022).
Mutations in the KRAS gene are the most common “drivers” of oncogenic alterations in NSCLC. Smoking is strongly associated with this tumor type, which is characterized by higher mutational loads and increased PD-L1 expression. Patients with KRAS mutations often present co-mutations in genes such as STK11, KEAP1, CDKN2A/B, and TP53, which define a subgroup of tumors that are particularly challenging to treat (Skoulidis and Heymach, 2019; West et al., 2022). In our sample, we identified KRAS mutations in 15% of individuals, which we attribute to a lower prevalence of heavy smokers compared with recent Brazilian (ranging from 24% to 27%) and worldwide data (24.5%) (Freitas et al., 2020; Mascarenhas et al., 2021; West et al., 2022). Mutations in the KRAS gene, particularly when accompanied by co-mutations in STK11/LKB1 and KEAP1, are associated with poor prognosis and low survival rates, even in patients with positive PD-L1 expression. This holds true despite the availability of treatment regimens that combine chemotherapy, antiangiogenic therapy, and anti-PD-1/PD-L1 immunotherapy (Schoenfeld et al., 2020; West et al., 2022). Additionally, we observed a significant association between mutations resulting in the loss of function of the MTAP and CDKN2A/B genes. Mutations in CDKN2A/B were found to co-occur with MTAP mutations in 98% of cases, and in our sample, this association was present in 100% of individuals (Awaya et al., 2004; Tam et al., 2013).
A mutation in PD-L1 was observed with significantly different expression levels compared with TCGA data in our population, potentially predicting a worse prognosis. This mutation has been widely discussed as a crucial therapeutic target and biomarker for treatment response (Ahn and Nagasaka, 2023; Gandhi et al., 2018; Rittmeyer et al., 2017). Previous studies have shown an association between PD-L1 expression and mediastinal lymph node infiltration in surgical specimens, with promising results in preoperative anti-PD-1 therapy trials (Osoegawa et al., 2023). One study found that 61% of samples were negative for PD-L1, while 39% showed positive tumor proportion score (TPS), with 18% demonstrating hyper-expression and 21% showing moderate expression (Rizvi et al., 2019). Another study confirmed these results in patients with adenocarcinoma, reporting that 66.6% had a TPS of <1% (Schoenfeld et al., 2020). In both studies, high PD-L1 expression was significantly more common in metastatic disease compared with primary tumor biopsies (P < 0.001).
The EXPRESS trial, the most extensive study on PD-L1 IHC expression (clone 22C3), involved 2,435 patients. It found that 48% of patients had negative expression, 30% had moderate expression, and 22% exhibited hyper-expression, with no significant differences based on biopsy origin (Dietel et al., 2019). In our study, we observed high IHC expression of PD-L1: 52.5% of the sample had a TPS of <1%, 14.6% exhibited high expression, and 32.7% had a TPS between 1% and 49%. Our data are consistent with those published in a recent single-center Chinese series (Liu et al., 2023). However, a statistically significant difference was observed between our sample and the worldwide literature (P = 0.04). The most substantial evidence currently published in Brazil comes from a retrospective cross-sectional study by the Latin American Cooperative Oncology Group and the GBOT. Gelatti and others reported that 60.6% of adenocarcinomas had negative TPS, 23.2% had moderate expression, and 16.2% were hyper-expressors (Gelatti et al., 2020). Comparing the data from the two studies, no significant difference was demonstrated (P = 0.5162).
In the two publications from the CHECKMATE-227 study, ∼58% of patients underwent molecular profiling (Hellmann et al., 2018, 2019). In our cohort, 13% of individuals exhibited a high TMB, indicating that 87% had TMB values below 10 mutations per megabase. In comparison, Mascarenhas and others reported high TMB in only 5.5% of cases among 80.5% of patients assessed (Mascarenhas et al., 2021). Our analysis revealed an antagonistic statistical correlation between TMB and the presence of activating mutations, as well as between smoking and activating mutations. However, no significant association was found between biomarker status (BMR) and smoking history.
In silico functional analysis of the gene set in our population demonstrated enrichment in pathways involved in signal transduction, transcription regulation, and cellular communication. These alterations were predominantly associated with gain-of-function mutations, promoting aberrant signaling through pathways such as IFN-γ, MET, PDGF, IGF-1, and ErbB—highlighting a high degree of interaction and cross talk among these oncogenic drivers (Bontoux et al., 2023; Sompallae et al., 2023).
A recent Brazilian cohort study evaluating early-stage NSCLC identified EGFR mutations in 17.3% of patients. PD-L1 positivity was observed in 36.7% of cases, with significant associations found between PD-L1 expression, smoking history, and advanced disease stages. These findings emphasize the heterogeneity of PD-L1 expression in relation to EGFR status and underscore their clinical relevance for treatment planning (Alves Pinto et al., 2022).
The interaction between PD-L1 expression and TP53 mutations has been increasingly recognized. Studies have shown that TP53 mutations are linked to elevated PD-L1 expression. In particular, KRAS-mutant tumors co-mutated with TP53 exhibit heightened PD-L1 expression, suggesting a synergistic mechanism that enhances tumor immunogenicity (Lamberti et al., 2020). These insights reinforce the growing emphasis on personalized medicine in NSCLC, highlighting the value of integrating molecular and immunological profiling to improve treatment outcomes.
It is important to acknowledge potential limitations of our study, notably the recruitment of most patients from private medical centers (n = 123; 92.5%), which may introduce selection bias. Our sample includes a higher proportion of non-smokers and individuals with metastatic disease. In contrast, comparative data from TCGA primarily derive from surgical specimens (80%), with a predominance of treatment-naive smokers, which may influence the observed genomic differences.
The genomic characterization of patients with NSCLC from Paraná reveals a distinct regional molecular signature, marked by a high prevalence of EGFR and TP53 mutations, along with increased PD-L1 expression. Previous research has shown that EGFR mutations, particularly in exons 19 and 21, are more frequent in certain populations and critically influence responsiveness to TKIs (Lynch et al., 2004; Paez et al., 2004). TP53 mutations, central to genomic stability and tumor suppression, have been linked to more aggressive tumor biology and treatment resistance (Kandoth et al., 2013). Furthermore, PD-L1 overexpression—a key immune checkpoint regulator—has clinical relevance in the context of immunotherapy, potentially predicting responses to checkpoint blockade (Herbst et al., 2014). Altogether, these findings highlight the significance of identifying regional genomic variations to tailor personalized therapeutic approaches and improve clinical outcomes in patients with NSCLC from Paraná and other genetically comparable populations.
Footnotes
Acknowledgment
The authors thank the Brazilian National Council for Scientific and Technological Development (grant 305335/2021-9 to C.P.) for supporting this study.
Authors’ Contributions
C.P. and G.L.L. contributed to the study design, conceptualization, data analysis, and the initial draft of the article. The other authors were involved in critically reviewing and revising the article.
Data Availability
Data will be available upon a reasonable request.
Author Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding Information
The authors declare that the research was conducted in the absence of any financial relationships.