
Abstract
Purpose:
Tafluprost is a novel prostaglandin F2α-receptor agonist shown to lower intraocular pressure (IOP) in healthy humans and patients with elevated IOP. We investigated the efficacy, safety, and tolerability of tafluprost 0.0015% compared with latanoprost 0.005% in patients with primary open-angle glaucoma, exfoliation glaucoma, or ocular hypertension.
Methods:
This was a randomized, double-masked, active-controlled, parallel-group, multinational, and multicenter phase II study. Patients received either tafluprost 0.0015% (n = 19) or latanoprost 0.005% (n = 19), both once daily. The extent and duration of action of the IOP-lowering effects at Day 42 and Day 43 were the primary efficacy endpoints. Efficacy and safety parameters were analyzed throughout.
Results:
Maximum IOP reduction was achieved by Day 7 and was sustained until Day 42 in both groups (mean [standard deviation] change from baseline −9.7 [3.3] mm Hg for tafluprost and −8.8 [4.3] mm Hg for latanoprost). The overall treatment group difference was 0.17 mm Hg (95% confidence interval −1.27 to 1.61; P = 0.811). The IOP-lowering effect was maintained for ≥24 h after the last dose in both groups. Most adverse events were ocular and were similar in frequency and severity between groups. There were 3 severe adverse events, all ocular, and all in the tafluprost group (3/19 = 16%).
Conclusions:
Tafluprost and latanoprost have comparable effects on the extent, duration, and stability of IOP reduction, and are well tolerated in patients.
Introduction
G
There are currently several medications available to reduce IOP in glaucoma, including prostaglandins, β-adrenergic receptor antagonists (β-blockers), and α-adrenergic receptor agonists (α-agonists).5 Prostaglandin analogs have gained popularity because of their efficacy in lowering IOP in patients with open-angle glaucoma or ocular hypertension, with less systemic adverse events compared with some other medications, such as β-blockers and α-agonists.6
Tafluprost is a novel, synthetic, prostaglandin, fluoroprostaglandin (FP) receptor agonist associated with potent FP receptor binding.7–9 It has been shown to reduce the IOP in both normotensive and hypertensive monkeys10 and in healthy human volunteers.11 Findings from a phase III clinical study has shown that tafluprost was well tolerated and provided additional IOP reductions when used as adjunctive therapy in glaucoma patients receiving timolol therapy.12 Tafluprost has recently been approved for the treatment of glaucoma and ocular hypertension in some European countries.13
The objective of this study was to investigate the efficacy, safety, tolerability, and duration of action of tafluprost (0.0015%) compared with latanoprost (0.005%) eye drops in patients with primary open-angle glaucoma, exfoliation glaucoma, or ocular hypertension. The aim was to investigate the efficacy of the study medications at the end of the 6-week treatment period, up to 48 h after the last dose.
Methods
This was a randomized, double-masked, active-controlled, parallel-group, multinational, and multicenter phase II study in 38 patients with primary open-angle glaucoma, exfoliation glaucoma, or ocular hypertension. The study was conducted across 3 sites in Italy and Finland, and in accordance with the International Conference of Harmonisation Good Clinical Practice guidelines, the Declaration of Helsinki, and was applicable to the Independent Ethics Committee and regulatory requirements.
Study design
After an appropriate washout period, patients were randomized to receive either tafluprost 0.0015% or latanoprost 0.005% eye drops for 6 weeks. Randomization was carried out using randomly permuted blocks, separately for each country. Patients were randomized using Proc Plan, SAS® for Windows (version 8.2; SAS Institute Inc., Cary, NC). Patients received study medication until Day 41. There were 2 visits during the treatment period (Days 7 and 21) and another 2 visits after the final dose had been administered (Days 42 and 43).
Patients
Eligible patients were aged ≥18 years, had primary open-angle glaucoma, exfoliation glaucoma, or ocular hypertension (with an IOP of 22–34 mm Hg in at least one eye), provided written, informed consent, and were willing to follow the study protocol. Patients were excluded if they had any uncontrolled systemic disease, were pregnant, had undergone surgery in the previous 6 months (cardiovascular, respiratory, or ocular), had any known allergy or hypersensitivity to the study medications or their components (including benzalkonium chloride), or had any history of ocular disease other than glaucoma.
Treatments
Patients on prior glaucoma medication had a minimum washout period of ≥4 weeks for β-blockers or prostaglandin analogs; ≥3 weeks for α-agonists; and ≥5 days for carbonic anhydrase inhibitors and miotics. During the required washout period, brinzolamide (Azopt®; Alcon, Fort Worth, TX) run-in medication was used if necessary. Brinzolamide was stopped ≥5 days prior to the end of the required washout period, to allow a sufficient washout period from all IOP-lowering medications. Washout durations were in-line with those previously reported.14,15
After the washout period, each patient was treated with either tafluprost 0.0015% eye drops (Santen Oy, Tampere, Finland), containing 0.1 mg/mL benzalkonium chloride, or latanoprost 0.005% eye drops (Xalatan®; Pfizer, New York, NY), containing 0.2 mg/mL benzalkonium chloride. A 0.0015% dose of tafluprost was selected based on the study in healthy volunteers11 and data from phase II dose–response relationship studies [Santen Oy; data on file] that showed that the dose of 0.0015% was the optimal therapeutic concentration, that is the lowest concentration that reliably produced the maximal IOP-lowering response or the dose with best balance for efficacy and tolerability. Tafluprost and latanoprost were dosed once daily in the evening (at 8:00
Concomitant treatment with any medication that could have had an effect on the study results was prohibited during the study.
Endpoints
IOP. The primary efficacy endpoints were the extent and duration of action at the end of the 6-week treatment period using the IOP measurements at Days 42 and 43 (ie, measurements up to 48 h after the last dose), and the individual IOP fluctuations after the last treatment dose. The secondary efficacy endpoint was the IOP values at 8:00 am on Days 7, 21, and 42. The proportions of patients reaching prespecified IOP reductions (≥15%, ≥20%, ≥25%, and ≥30%) were also evaluated.
Diurnal IOP measurements were taken at 8:00 am, 12:00 pm, 4:00 pm, and 8:00 pm on Day 0 (baseline), and Day 42 (ie, 12, 16, 20, and 24 h after the last dose). Further IOP measurements were taken at 8:00 am and 8:00 pm on Day 43 (ie, 36 and 48 h after the last dose). IOP was also measured at 8:00 am on Days 7 and 21.
The primary evaluation of IOP was based on the “worse eye,” which was defined as the eye with the higher IOP at the 8:00
Adverse events. Safety endpoints included overall adverse events, best-corrected visual acuity, conjunctival hyperemia, biomicroscopy, fundus examination, ocular symptoms, overall drop discomfort, blood pressure, and heart rate.
All adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA; version 7.1). A description of the event, whether or not it was serious, the onset and duration, the frequency, the severity, the relationship to the study medication, the location (left or right eye, both eyes, not applicable), the action taken, and the outcome were also included.
Corrected visual acuity was measured at each study visit using an Early Treatment Diabetic Retinopathy Study (ETDRS) chart, and changes of at least 0.2 LogMAR scores were identified. Changes from baseline in conjunctival hyperemia were assessed at each post-screening visit using a severity grading (0 = none, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe) and picture sample charts, as previously described.16,17 A biomicroscopic examination—including the lids, conjunctiva, cornea, anterior chamber, iris, lens, and vitreous—was performed at each study visit and findings were graded (1 = mild, 2 = moderate, or 3 = severe). An examination of the fundus was performed at screening and the post-study visit and also graded (1 = mild, 2 = moderate, or 3 = severe). All ocular symptoms were evaluated at each study visit, excluding the post-study visit and the related visual analog scale severity scores (0–100) that were summarized. Overall drop discomfort was evaluated by patients on Days 7, 21, and 42 using a 4-point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe). Blood pressure and heart rate were measured at each study visit, except the post-study visit.
Statistical and analytical plans
All randomized patients who received at least one dose of study treatment and from whom at least one efficacy or safety (including tolerability) measurement was obtained after randomization were included in the intention-to-treat efficacy dataset or safety dataset, respectively.
The extent and duration of action at the end of the 6-week treatment period (IOP measurements up to 48 h after the last dose on Days 42 and 43) were evaluated using a repeated measurements analysis of variance (RMANOVA) model. A 95% confidence interval (CI) for the overall treatment effect (tafluprost − latanoprost) was calculated from the RMANOVA model. The results were further characterized using a random coefficients regression model. The stability of IOP measurements on Days 42 and 43 was evaluated by calculating univariate characteristics (coefficient of variation, minimum, maximum, and range) separately for each patient. These values were then summarized by treatment group with descriptive statistics in order to quantify the fluctuations. The changes from baseline in diurnal IOP at Day 42 and the 8
The evaluation of safety and tolerability was based on all treated eyes. Adverse events were presented in frequency tables for both patient and event count. Ocular and non-ocular events were reported separately. Visual acuity, conjunctival hyperemia, biomicroscopy, ocular symptoms, and fundus examination findings were presented using appropriate summary statistics, and overall drop discomfort was summarized descriptively.
The Fisher exact test was used to test for differences in categorical data, such as for baseline demographics and proportion of responders, and the t-test was used for continuous data. No prospective sample size calculation was done for this phase II study. However, the retrospectively calculated power with the 18 completed patients per group was 78%. The calculations were based on the RMANCOVA estimate of variability (standard deviation [SD] = 2.12 at Day 42), a clinically significant difference of 2 mm Hg and a 2-sided α-level of 0.05.
Results
Patient distribution
Of the 46 screened patients, 38 were randomized; 19 patients in each treatment group. A total of 36 patients completed the study, 18 in the tafluprost group and 18 in the latanoprost group. Two patients, one in each group, discontinued the study due to adverse events on Day 9 (their last study visit was Day 7; Fig. 1)

The flow chart of study patients distribution. Two patients, one in each group, discontinued treatment before Day 42 after reporting several adverse events (including conjunctival hyperemia) that were considered probably related to the study treatments.
Demographic and other baseline characteristics
Demographic and baseline characteristics are outlined in Table 1. There was greater proportion of female patients in the latanoprost group (16/19, 84.2%) compared to the tafluprost group (10/19, 52.6%). Overall, slightly over half of the patients had brown eyes (21/38, 55.3%), and the majority of patients in both groups had primary open-angle glaucoma. A total of 16/19 (84.2%) patients in both treatment groups reported use of any prior medication, of whom 9 patients in the tafluprost group and 11 patients in the latanoprost group reported prior use of ophthalmological treatment requiring washout.
P
aFisher exact test for categorical data (brown/other distribution tested in iris color), t-test for continuous data.
bWorse eye data.
Abbreviation: SD, standard deviation.
Efficacy
Level of IOP reduction. The full IOP-lowering effect was achieved by the 8:00
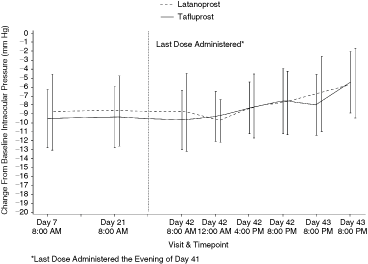
Mean of intraocular pressure (mm Hg; ± standard deviation) changes throughout the study at all time points. Data to the right of the vertical dotted line were all taken after the last dose administered in the evening of Day 41.
M
Extent, duration, and stability of IOP reduction. The 8:00
The mean IOP measurements over the last 2 days of the study were comparable between tafluprost and latanoprost (P = 0.143; RMANOVA treatment by time interaction; Fig. 2). The 2 treatments were similar with respect to the IOP during Days 42 and 43. The estimated overall treatment difference (tafluprost − latanoprost) in IOP values during Days 42 and 43 was 0.056 mm Hg (95% CI −1.497 to 1.608 mm Hg; P = 0.942; RMANOVA treatment effect).
Responder analysis. The responder analysis showed that the number of patients who achieved the target IOP values was comparable between the tafluprost and latanoprost groups. Tafluprost had a higher proportion of responders at all time points for all prespecified IOP reductions (≥15%, ≥20%, ≥25%, and ≥30%; Table 3). For example, at Day 42, more patients achieved a prespecified ≥20% IOP reduction at all time points with tafluprost compared with latanoprost (14/18 [77.8%] versus 9/18 [50.0%], respectively).
P
aFisher exact test.
Peak-trough IOP. The morning IOP reductions at 8:00
Safety
Overall, approximately one-third of the patients in each group reported an adverse event; there was no increase in the number of adverse events throughout the 42 days of treatment in either treatment group. A total of 17 adverse events (13 ocular and 4 non-ocular) were reported by 6 patients in the tafluprost group, whereas 23 adverse events (17 ocular and 6 non-ocular) were reported by 7 patients in the latanoprost group. The only non-ocular adverse event that was considered related to the study treatment was a single case of headache in the latanoprost group.
A total of 3 adverse events were considered severe, all of which occurred in the tafluprost group (2 photophobias and 1 eye pruritus). One serious adverse event (macular hole/cyst) was detected at the post-study visit (Visit 7) in a patient who received tafluprost during the study, but was switched to latanoprost after the evening of Day 43. The event was considered unlikely to be related to the study medication.
Two patients, one in each group, discontinued the study prematurely due to adverse events. Both patients reported several adverse events (including conjunctival hyperemia) that were considered probably related to the study treatments.
There were no deaths or variations in blood pressure or heart rate in either treatment group, and overall drop discomfort was similar between the treatment groups.
Ocular safety
Most of the adverse events were ocular (30/40 [75%]) and were evenly distributed across the 2 treatment groups (Table 4). Overall, 4 of 19 (21.1%) patients in each group reported drop discomfort (3 mild and 1 moderate in the tafluprost group, and 2 mild and 2 moderate in the latanoprost group).
S
A single adverse event was counted once for each patient by maximum severity.
Abbreviation: MedDRA, Medical Dictionary for Regulatory Activities.
For visual acuity, the LogMAR scores remained stable throughout the study in both treatment groups. No differences between the treatment groups were detected during the biomicroscopic examination of the lids, cornea, anterior chamber, iris, lens, and vitreous humor. The ocular symptoms (irritation/burning/stinging, foreign body sensation, tearing, itching, photophobia, dryness, and other) were also similar in the tafluprost and latanoprost groups.
In both treatment groups, most of the patients had no conjunctival hyperemia during the study. The largest increases in mean conjunctival hyperemia from baseline were seen at Day 7. The mean increases that were very mild were classified as less than grade 1. The largest increases, defined as change of 2 grades from baseline, were experienced by 3 patients in both treatment groups. After Day 7, conjunctival hyperemia was more prominent in the latanoprost group (Fig. 3). By Day 43, conjunctival hyperemia was back at baseline levels for the tafluprost group.
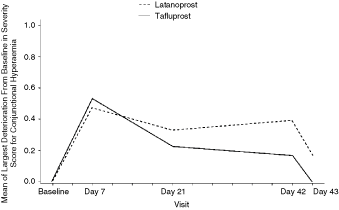
Mean (± standard error [SE]) of largest increase from baseline in conjunctival hyperemia severity score. Note: Differences were not statistically significant.
Discussion
Our study is the first to investigate the efficacy, safety, and tolerability of tafluprost compared with latanoprost in patients with primary open-angle glaucoma, exfoliation glaucoma, or ocular hypertension. In this pilot phase II study, the extent, duration, and stability of IOP reduction of tafluprost and latanoprost were comparable.
The IOP reductions achieved with both tafluprost and latanoprost in this study were maintained for over 24 h after the last dose was administered for both agents. This is in agreement with a previous study that investigated the duration of IOP-lowering effect 24, 36, and 48 h after a single latanoprost administration.14 The study found that the reduction in IOP was maintained over 24 h, and was still present, but less pronounced, after 48 h.14
For both drugs, the mean IOP reductions obtained in this phase II study are in line with those seen in previous studies with latanoprost.18–21 Likewise, for both drugs, the peak and trough IOP reductions observed in this study were also consistent with previous studies. A meta-analysis of several randomized studies compared the peak and trough IOP reductions for various anti-glaucoma medications. The peak and trough IOP reductions (95% CI) for latanoprost were: −31% (−33% to −29%) and −28% (−30% to −26%), respectively.21 Both peak and trough values for latanoprost in this present study are within the range previously observed in the meta-analysis; peak range −33% to −32.3%, and trough −29.9%. The peak (means −35.6%, −34.3%, and −35.9% on Days 7, 21, and 42, respectively) and trough (mean −29.2% for tafluprost) IOP reductions observed with tafluprost in the present phase II study are similar with those reported with other prostaglandin analogs within the meta-analysis.21
In the present study, the IOP reductions produced by tafluprost were comparable with latanoprost, within the power of this study to detect differences of ∼2 mm Hg. This confirmed that tafluprost is a promising therapeutic agent, and accordingly larger phase III clinical trials were started.
The present study showed that tafluprost was well tolerated in patients with glaucoma or ocular hypertension. The safety profile observed with tafluprost was comparable with that observed with the commercially available prostaglandin latanoprost. There was only one serious adverse event that was mild and unrelated to medication, and 3 severe adverse events, all ocular, and all in the tafluprost group (3/19 = 16%; one eye pruritus, 2 photophobias).
The rates of conjunctival hyperemia were also comparable between tafluprost and latanoprost. Conjunctival hyperemia is one of the most common adverse events associated with glaucoma therapy and it has been linked to premature treatment discontinuation.22 The rates of hyperemia vary between the currently available prostaglandin analogs,22 and it has been hypothesized that the differences in conjunctival hyperemia may explain why discontinuation rates vary between the prostaglandin analogs.23
Tafluprost is a new prostaglandin analog for the treatment of glaucoma and ocular hypertension. Based on this phase II study, tafluprost has a good efficacy and safety profile and, therefore, more extensive, larger-scale, phase III trials have been initiated.
Footnotes
Acknowledgment
The authors would like to thank Teppo Huttunen for his statistical contributions to this study.
Author Disclosure Statement
C.E.T. has received departmental research funding, and has been an advisor for Merck, Pfizer, Santen Oy, Allergan, and Glaukos. A.R. is an employee of Santen Oy, Finland. For M.P. no competing financial interests exist. H.U. has been a consultant for and has received research funding from Santen Oy and Pfizer.
The study was sponsored by Santen Oy and all study related material including, for example study protocol and study medication, was prepared by the sponsor. Investigators were contractors to Santen (responsible for the study conduct). The authors received editorial/writing support in the preparation of this manuscript, funded by Santen Oy. The authors are fully responsible for text, data, and editorial decisions for this manuscript.