
Abstract
Abstract
Purpose:
β-Adrenergic receptor (βAR) agonists reduce intraocular pressure (IOP) by increasing aqueous outflow through the trabecular meshwork (TM). However, although this effect is well established, the specific signaling mechanisms responsible are less clear. To address this, the current study examined βAR signaling in primary human trabecular meshwork cells (HTMCs), specifically, focusing on the effect of βAR activation on the extracellular signal regulated kinases 1/2 (ERK).
Methods:
HTMCs were cultured and assessed for βAR expression by both immunofluorescence and reverse-transcription–polymerase chain reaction. The effect of βAR activation on ERK phosphorylation was examined in these cells by In-Cell Western™ analysis. Pharmacological approaches were used to characterize the mechanism of the βAR effect on ERK.
Results:
Treatment of HTMCs with the nonselective βAR agonist, isoproterenol (ISO), decreased the basal phospho-ERK (pERK) level, through actions at the β2AR. The response was insensitive to pertussis toxin (PTx), but pretreatment with cholera toxin (CTx) resulted in a reversal of the response, such that ISO treatment instead increased pERK, thus implicating Gαs in the inhibitory pERK response. The adenylyl cyclase activator, forskolin, also decreased pERK, suggesting the involvement of adenylyl cyclase and cAMP, whereas a protein kinase A (PKA) inhibitor, H-89, blocked both ISO and forskolin-mediated pERK inhibition in HTMCs. Finally, a closer examination of the pERK increase generated in the presence of CTx demonstrated that it was also insensitive to PTx, and appeared to have differing rank orders of efficacy for various βAR agonists compared with the inhibitory pERK pathway in HTMCs.
Conclusion:
A novel β2AR-signaling pathway leading to a decrease in pERK, which was dependent on Gαs, cAMP, and PKA, was identified in HTMCs. A competing β2AR signaling pathway resulting in increased pERK was also identified. Since βAR effects on aqueous humor (AH) outflow have been linked to cAMP, and inhibition of ERK in TM cells has recently been suggested as increasing AH outflow, our findings suggest that the inhibitory β2AR-pERK pathway likely represents the mechanism by which βAR agonists decrease IOP. The presence of a competing β2AR-ERK activation pathway in the same cells suggests that this is an ideal system for the development and validation of functionally selective β2AR agonists.
Introduction
The importance of β-adrenergic receptor (βAR) signaling in the regulation of IOP has been recognized for some time. βAR antagonists are currently a front-line treatment for glaucoma based on their IOP-lowering actions, which derive primarily from a decrease of AH secretion.4,5 In addition to the effect of βAR antagonists, βAR agonists also reduce IOP, but do so by increasing AH outflow in the TM.6,7 Although the IOP-lowering effect of βAR agonists on the TM has been suggested to involve the activation of adenylyl cyclase, resulting in cAMP production,6,8 a comprehensive examination of βAR signaling in these cells, including the contribution of additional βAR-coupled signaling pathways, and how they may contribute to the βAR agonist effect on AH outflow has not been conducted.
One βAR signaling pathway that has received considerable attention in other cell types and tissues is the extracellular signal regulated kinase 1/2 (ERK) pathway. Extensive studies utilizing both heterologous expression systems and native cells have demonstrated that βAR activation can increase ERK phosphorylation through many different signaling mechanisms, including via Gαs, Gαi, Gβγ, and β-arrestin-mediated pathways.9–12 Despite this, there is also evidence in certain cell types that βAR signaling may instead lead to a decrease in ERK phosphorylation, occurring through both cAMP-dependent and independent pathways.13–16 In addition, several recent studies have linked the activity of ERK to both the synthesis and degradation of extracellular matrix (ECM) proteins by the TM, thus suggesting that ERK signaling pathways may play a critical role in regulating AH outflow in this tissue.17–19
Given the multiplicity in βAR-pERK signaling responses, and the importance of ERK and the βARs in the regulation AH outflow and IOP within the TM, clarifying βAR-pERK signaling mechanisms within TM may identify additional approaches of targeting these receptors, or their signaling pathways, for glaucoma therapeutics. Therefore, the current study examined βAR-pERK signaling in primary human trabecular meshwork cells (HTMCs). Our results provide evidence for competing β2AR-pERK signaling pathways in human TM cells, thereby leading to either a decrease or an increase in pERK. Our findings suggest that it is likely that the inhibitory pERK pathway is responsible for the AH outflow and the IOP effects of βAR agonists, and therefore, we propose that functionally selective βAR agonists, selectively activating this pathway, may represent an alternative, more efficacious means of targeting these receptors to reduce IOP.
Methods
Cell culture
Primary HTMCs were obtained from ScienCell Research Laboratories, Carlsbad, CA. Primary HTMCs were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C, and in a 5% CO2 atmosphere. All experiments on HTMCs were carried out between passages 3 and 10.
The TM5 human TM-derived cell line was generously provided by Alcon laboratories. TM5 cells were maintained in high-glucose DMEM supplemented with 10% FBS at 37°C and in a 5% CO2 atmosphere. All experiments on TM5 cells were carried out between passages 5 and 15.
Phase contrast microscopy
HTMCs were plated in plastic dishes and maintained in culture media for 24–48 h. Cells were imaged while in culture media by using a Nikon inverted microscope and an Infinity3 cooled-CCD camera (Luminera Corporation).
Immunofluorescence and confocal microscopy
HTMCs were plated onto glass cover slips and maintained for 24–48 h in culture media before fixation with 100% ice cold methanol. After washing the cover slips with phosphate-buffered saline (PBS), cells were blocked with 1% bovine serum albumin (BSA) in PBS for 90 min at room temperature. Cover slips were incubated overnight at 4°C with an anti-β2AR primary antibody (Abnova). After washing with PBS, cover slips were incubated with a Cy3-conjugated anti-mouse immunoglobulin G (IgG) secondary antibody, 1:500 (Jackson ImmunoResearch Laboratories, Inc) for 1 h at room temperature, then washed with PBS before being mounted on slides using the Fluorsave reagent (Calbiochem).
Slides were imaged by using a Nikon Eclipse E800 microscope fitted with the D-Eclipse C1 confocal system (Nikon Canada Inc.). Cy3 was imaged by using a 543 nm He-Ne lazer (JDS Uniphase). All images were taken by using a 100×oil immersion objective, with the total magnification represented by a scale bar on each image.
RNA isolation and reverse-transcription-polymerase chain reaction
RNA was isolated from HTMC and TM5 cells by using an RNeasy kit (Invitrogen Canada Inc.) according to the manufacturer's protocol. The purity and concentration of the resulting RNA was assessed first by measuring the A260/280 ratio of the samples. Reverse-transcription polymerase chain reaction (RT-PCR) was then carried out to demonstrate the expression of adrenergic receptor or myocilin transcripts. Primers used were as follows: β1AR forward 5′-AAAATCGATCATCGTGGCTC-3′ and reverse 5′-GGTTTGCCCTACACAAGGAA-3′; β2AR forward 5′-GAGCACAAAGCCCTCAAGAC-3′ and reverse 5′-TGATATCCACTCTGCTCCCC-3′; myoclin forward 5′-CTTGGACCAGGCTGCCAGGC-3′ and reverse 5′-TCGGGTCTGGGGACACTGGC-3′; and β-actin forward 5′-AGCGAGCATCCCCCAAAGTT-3′ and reverse 5′-GGGCACGAAGGCTCATCATT-3′. The RT-PCR products were run on a 2% agarose gel and visualized under UV light with a Kodak 290 docking station. All PCR products obtained were verified based on expected amplicon size, and in all cases, the obtained product matched the predicted produce size.
In-cell western analysis
Phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK) and cyclic AMP response element binding protein (CREB) was assessed by using a modified in-cell western protocol as previously reported.20,21 For this, cells were plated in 96-well plates and cultured 24–72 h until confluent. Cell culture medium was then replaced with FBS-free DMEM, and cells were maintained for 24 h before experiments. Cells were treated as indicated in FBS-free DMEM at 37°C, 5% CO2 before fixation for 1 h with 4% paraformaldehyde.
To carry out the In-Cell Western™ assay, cells were first washed with PBS, then permeabilized for 1 h at room temperature with 0.1% triton X-100 in PBS. After washing again with PBS, nonspecific binding sites were blocked by incubating for 90 min at room temperature with 1% BSA in PBS supplemented with 0.1% tween-20 (PBST). Cells were then incubated overnight at 4°C with either a rabbit anti-phospho ERK primary antibody (Santa Cruz Biotechnology Inc), diluted 1:200; or a goat anti-phospho-CREB primary antibody (Santa Cruz Biotechnology Inc), diluted 1:200; each in PBST containing 1% BSA. Cells were washed with PBST, before being incubated for 1 h at room temperature with secondary antibodies diluted in PBST containing 1% BSA: IR800CW conjugated anti-rabbit IgG raised in donkey (Rockland Immunochemicals), diluted 1:800 for pERK; or with Alex Fluor 680 conjugated anti-goat IgG raised in donkey (Invitrogen Canada Inc), diluted 1:500 for pCREB. Cells were then washed thrice with PBST, before being incubated with total ERK and total CREB antibodies. For ERK, a goat anti-total ERK2 antibody (Santa Cruz Biotechnology Inc) was diluted 1:200 in PBST containing 1% BSA and incubated for 1 h at room temperature. In CREB experiments, a rabbit anti-total CREB primary antibody (Santa Cruz Biotechnology Inc) was diluted 1:200 in PBST and exposed overnight at 4°C. Cells were washed in PBST, then incubated in secondary antibodies diluted in PBST with 1% BSA for 1 h at room temperature: Alex Fluor 680 conjugated anti-goat IgG raised in donkey diluted 1:800 for ERK2; or IR800CW conjugated anti-rabbit IgG raised in donkey diluted 1:800 for CREB. Plates were then washed 4 times with PBST, twice with PBS, and once briefly with dH2O, before patting dry against a paper towel and allowing the plate to air-dry overnight.
Plates were scanned to measure their fluorescent emission from the IRDye800CW and Alexa Fluor680 conjugated antibodies by using the Odyssey infrared imaging systems 800 and 700 nm channels, respectively (Li-Cor Biotechnology). To convert these data to relative pERK and pCREB values, background fluorescence was first determined and then subtracted by using wells of the plate that received only the secondary antibodies. The ratio of the background-subtracted pERK/ERK2 or pCREB/total CREB signals were then determined for each well and normalized to the ratios obtained from the appropriate vehicle or untreated wells, to obtain relative pERK or pCREB values. All experiments were repeated multiple times, and within each experiment, each condition was repeated in 2–8 wells.
Materials
Pertussis toxin (PTx was from EMD Chemicals, ICI 118,551 (ICI) ((±)-erythro-(S*,S*)-1-[2,3-(Dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride), CGP 20712 (CGP) (1-[2-((3-Carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol dihydrochloride), and SR 59230A (1-(2-Ethylphenoxy)-3-[[(1S)-1,2,3,4-tetrahydro-1-naphth alenyl]amino]-(2S)-2-propanol hydrochloride) were from Tocris Bioscience. FBS was from PAA laboratories Inc. All other chemicals and reagents were from Sigma-Aldrich Canada Ltd.
Statistical analysis and curve fitting
All data are presented as mean±s.e.m. Statistical analysis and curve fitting of the data were performed by using Graphpad Prism version 5.0b for Mac OSX (GraphPad Software Inc.). To fit data to dose-response curves, vehicle treatments were plotted at a concentration equal to one or one and a half log units less than the lowest drug treatment concentration; then, data were fit to a sigmoidal dose-response curve with a variable slope. Unless otherwise indicated, statistical significance was determined: for curve fits by the F test comparing global fits; by t-test when comparing means of only 2 groups; or by one-way, or 2-way ANOVA, as appropriate, when comparing the means of multiple treatment groups. Tukey's post-hoc analysis was used to determine differences among groups for one-way ANOVA, whereas Bonferroni's post-hoc analysis was used for 2-way ANOVA. P<0.05 was considered statistically significant.
Results
βAR expression and myoclin induction in HTMCs
Primary HTMCs are adherent in culture, and have a narrow, elongated spindle-shaped morphology (Fig. 1A). Immunofluoresence labeling demonstrates that these cells express β2AR when grown in culture (Fig. 1B). The expression of the β1AR and β2AR transcripts in HTMCs were compared with their expression levels in TM5 cells by RT-PCR (Fig. 1C). Although there was very little or no detectable transcript expression for the β1AR in either the TM5 or HTMCs, both cell types were found to express the β2AR transcript. A control reaction using primers for the ubiquitously expressed β-actin served as a positive control for the RT-PCR reaction. These findings suggest that primary HTMCs, similar to TM5 cells, express β2AR, but not β1AR when maintained under our cell culture conditions. To further demonstrate that the cells used in this study were, in fact, of TM origin, we demonstrated that treatment of either TM5 cells or HTMCs with dexamethasone resulted in induction of myocilin RNA expression (Fig. 1D). Dexamethasone-induced expression of myoclin is well reported for TM cells, 22 and the fact that induction was observed in both TM5 cells and HTMCs indicates that both cell types are of TM origin.
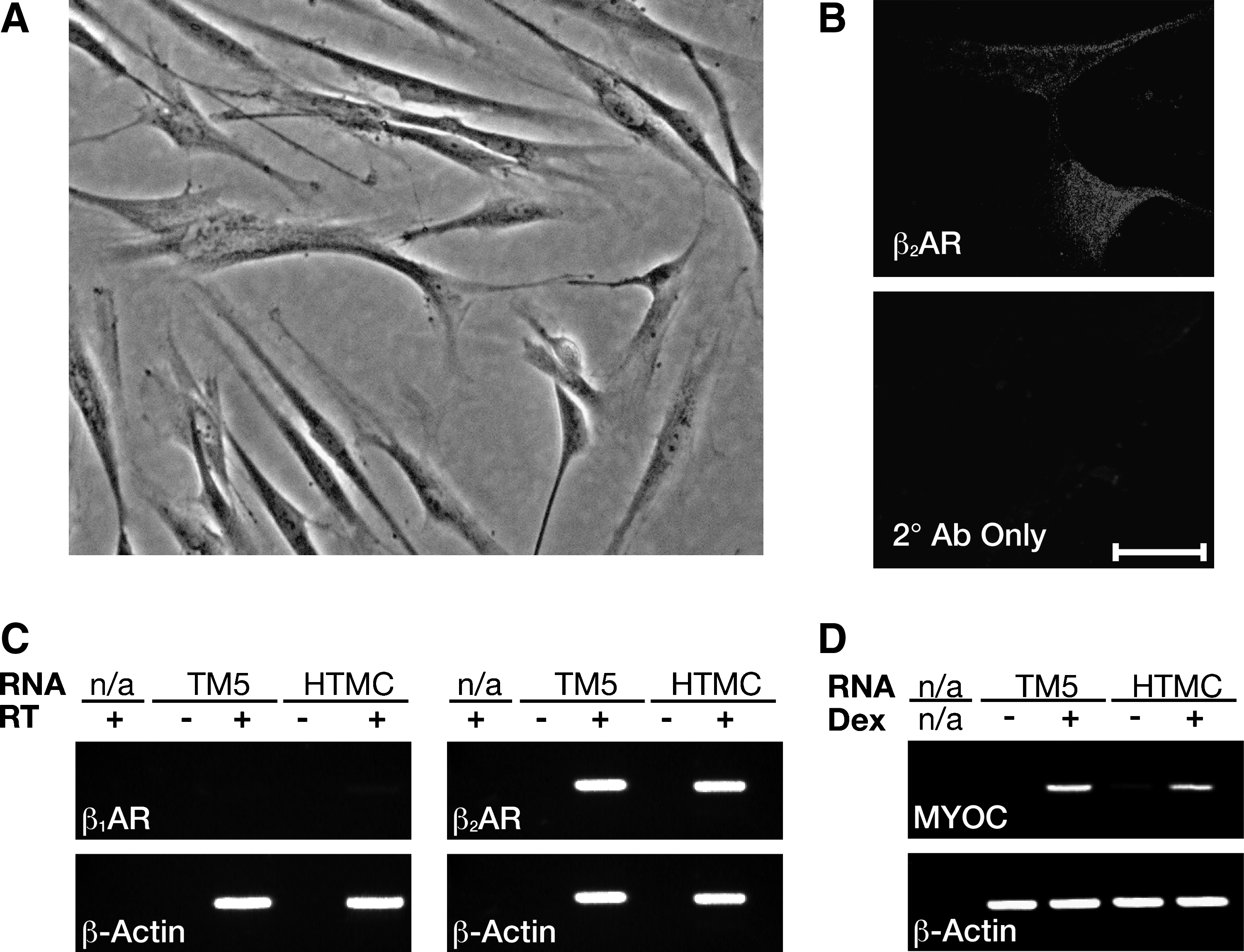
Primary HTMCs express β2AR and show induction of myocilin expression after treatment with dexamethasone.
To assess the functional expression of β2AR in HTMCs, the ability of the nonselective βAR agonist isoproterenol (ISO) to increase the level of phosphorylated CREB (pCREB) was assessed (Fig. 2A). ISO treatment of HTMCs resulted in a dose-dependant increase in pCREB, with a maximum 21%±2% increase in the pCREB level and a pEC50 of 8.5±0.2. To confirm that this ISO-pCREB response was mediated by β2AR, HTM cells were treated with ISO in the absence or presence of pretreatment with the β2AR selective antagonist ICI (Fig. 2B). In these experiments, ICI significantly attenuated the ISO-pCREB response at 10 (P<0.01), 30 (P<0.001), and 100 nM (P<0.001). These observations confirm that HTMCs functionally express β2AR, and that this receptor is coupled to an increase in the levels of pCREB.

β2AR activation in primary HTMCs leads to an increase in pCREB.
β2AR activation decreases pERK in HTMCs
Having demonstrated functional β2AR pCREB signaling in HTMCs, βAR pERK signaling was next examined in these cells. In primary HTMCs, surprisingly, ISO treatment resulted in a dose-dependant decrease in the basal pERK level (Fig. 3A). This ISO response produced a * 21%±4% reduction in the pERK level of these cells, and had a pEC50 of 7.5±0.4. Since activation of the βARs is often associated with an increase and not a decrease in pERK,11,23,24 ISO-pERK signaling was also assessed in a separate HTM-derived cell line (TM5 cells) to confirm that the observed decrease in pERK was not an anomalous effect unique to these particular primary cells (Fig. 3B). Similarly, in the TM5 cell line, as in the primary HTMCs, ISO treatment resulted in a significant reduction in the basal pERK level (P<0.001). Interestingly, in the TM5 cell line, the ISO response was actually even more pronounced, resulting in a maximum 52%±4% reduction from the baseline pERK level, compared with only the 21%±4% reduction observed in the primary HTMCs. This does not appear to reflect different levels of β2AR expression in the TM5 cells relative to the HTMCs, given that similar transcript levels were observed for β2AR in these 2 cell types (Fig. 1C). Instead, it is likely that a different expression of downstream effectors accounts for the larger response in TM5 cells.
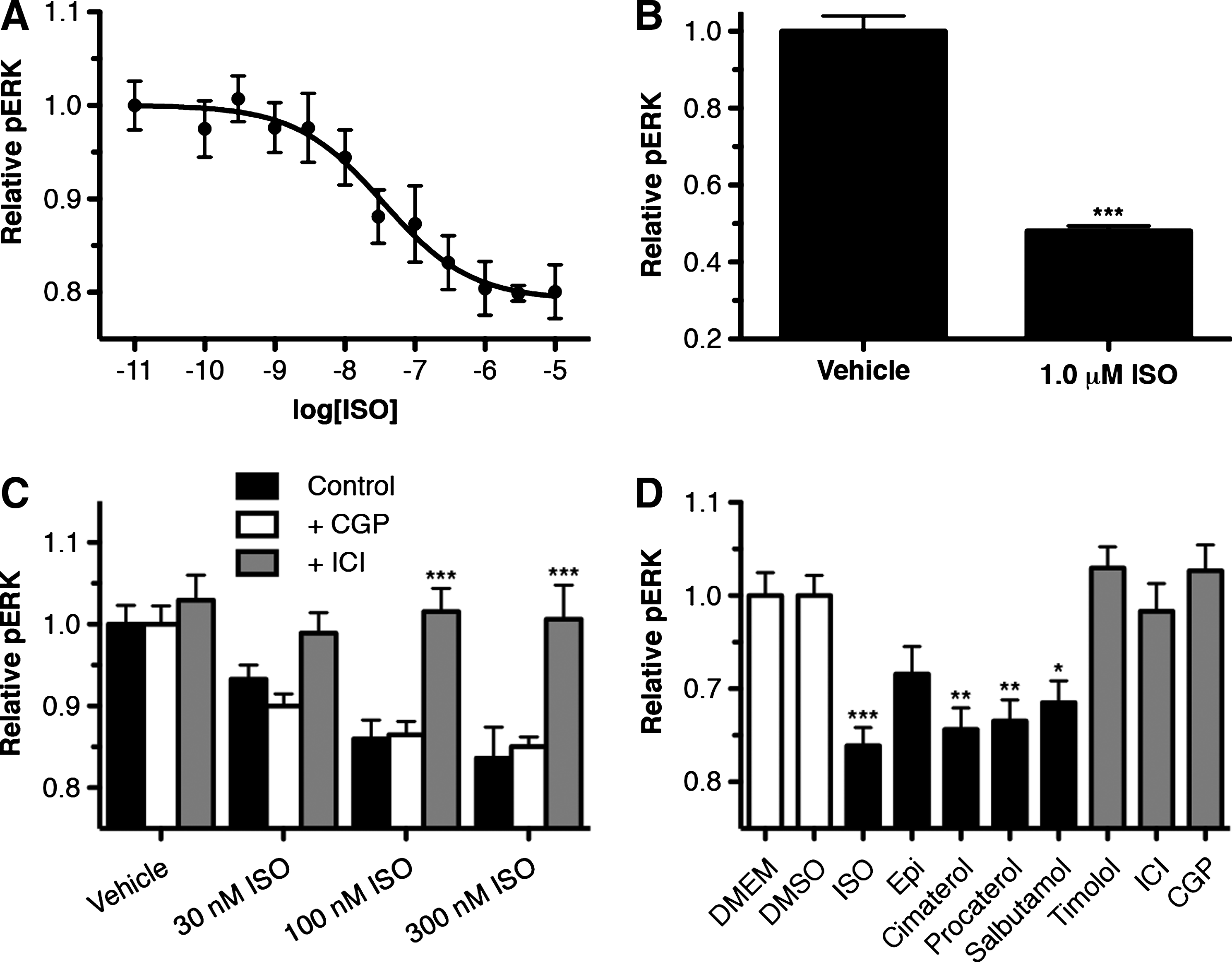
β2AR activation in HTMCs leads to a decrease in the basal pERK level of these cells.
Further investigation of the ISO-pERK reduction in primary HTMCs demonstrated that the ISO-response (100 and 300 nM) was significantly reduced by pretreatment with the β2AR selective antagonist ICI (P<0.001), but was unaffected by the β1AR antagonist CGP; thus confirming that the ISO-mediated reduction in pERK in HTM cells is via β2AR and not β1AR.
To assess whether there is functional selectivity in the β2AR-pERK response of HTMCs, the ability of various βAR ligands to affect pERK in these cells was examined (Fig. 3C). In these experiments, all βAR agonists tested, except epinephrine, produced significant decreases in pERK. Specifically, the nonselective agonist ISO produced the largest reduction of 16%±3% (P<0.001), followed by the nonselective agonist cimaterol, which produced a reduction of 14%±3% (P<0.01). The 2 β2AR-selective agonists, procaterol and salbutamol, each produced slightly smaller reductions of 13%±3% (P<0.01) and 11%±3% (P<0.05), respectively. Epinephrine, which is an endogenous adrenergic agonist for all adrenergic receptor subtypes and is typically associated with greater activity at β2AR than β1AR, did not produce a significant effect. However, epinephrine still appeared to produce a small reduction (8%±4%) in the pERK level of primary HTMCs. All 3 βAR antagonists tested, when applied on their own, had no significant effect on the basal pERK levels in these cells (P>0.05). Together, these findings indicate that activation of β2AR in HTM cells reduces the pERK levels in these cells, albeit with differences in efficacy based on the specific β2AR agonist used.
β2AR decreases pERK in HTMCs by activation of Gαs
The specific mechanism behind the β2AR-mediated decrease in pERK was then examined, by first considering which G proteins might contribute to the effect. To assess the role of Gαi/o in the response, ISO-pERK dose-response curves were generated in HTMCs in the absence or presence of pretreatment with PTx, a selective inhibitor of Gαi/o signaling (Fig. 4A). The dose-response curve was found not to be significantly different in the absence or presence of PTx (P>0.05), thus suggesting that Gαi/o is not involved in the β2AR-pERK response. To examine a potential role for Gαs in the response, the ISO-pERK dose response was examined in the absence or presence of long-term cholera toxin (CTx) pretreatment to desensitize Gαs (Fig. 4B). In this case, CTx pretreatment significantly altered the ISO-pERK dose-response curve (P<0.001). Specifically, not only did CTx pretreatment completely block the ISO-mediated decrease in pERK, but also after CTx pretreatment, ISO actually produced a dose-dependant increase in pERK. This ISO-mediated increase in pERK had an EMax 17% over basal with a pEC50 of 7.9±0.2. These observations suggest that not only is the HTMC β2AR-mediated decrease in pERK the result of β2AR-Gαs coupling, but also that blocking this pathway unmasks an underlying signaling pathway that results in an increase in pERK in these cells. To next establish whether this increase in pERK in the presence of CTx is the result of β2AR-Gαi/o coupling, ISO-pERK dose responses were generated in HTMCs in the absence or presence of pretreatment with a combination of CTx to produce the ISO-mediated increase in pERK, and PTx to inhibit Gαi/o signaling (Fig. 4C). In the absence of CTx and PTx, ISO produced a 15%±2% reduction in pERK, whereas in the presence of CTx and PTx, ISO resulted in a 17%±2% increase in pERK. Both responses had similar pEC50 values of 7.7±0.2 and 8.3±0.2, for the control and CTx/PTx treated HTMCs, respectively (P>0.05). Given that ISO treatment resulted in an increase in pERK in the presence of both CTx and PTx, these results indicate that this pERK increase is independent of both Gαs and Gαi/o coupled pathways.
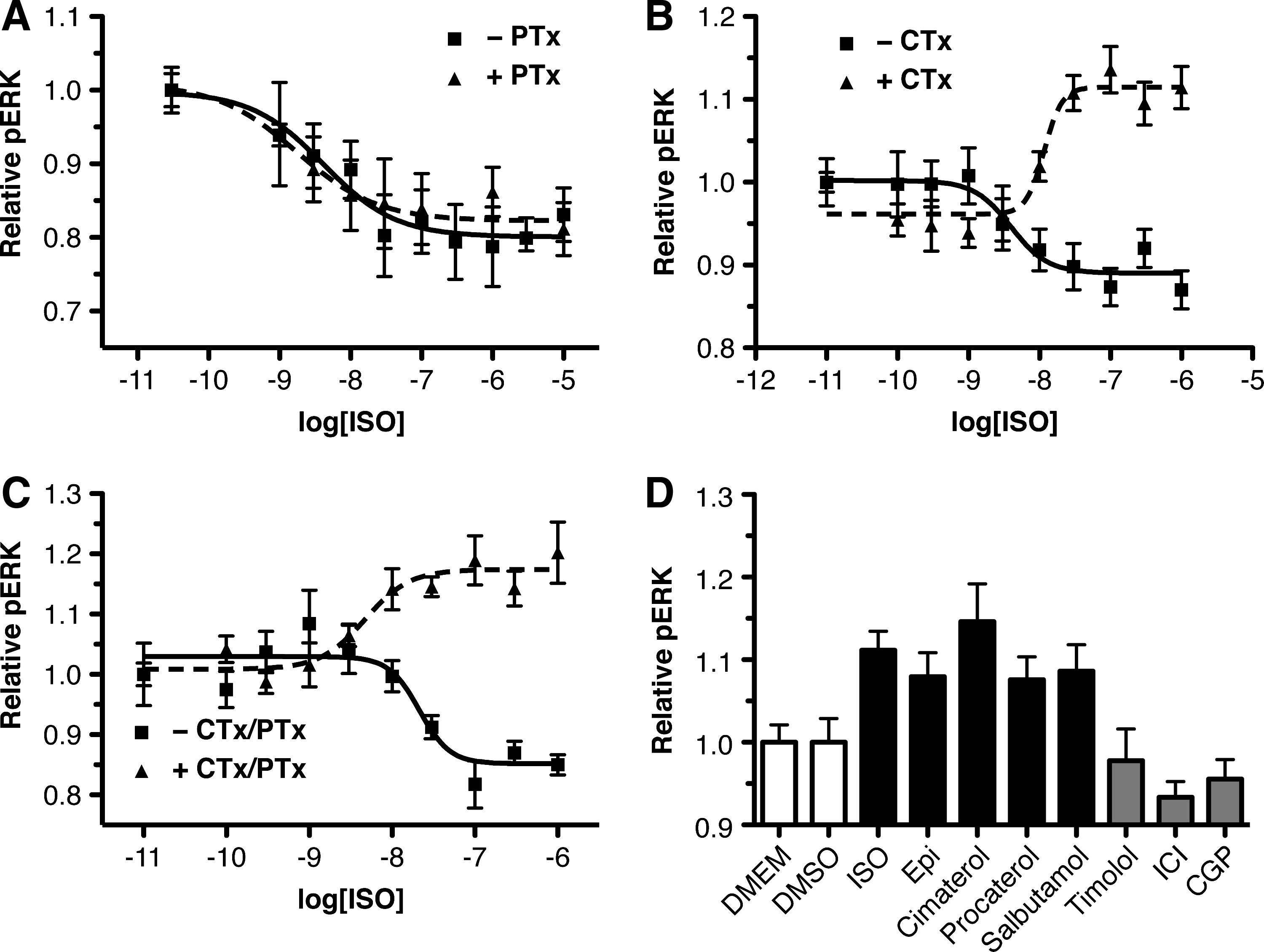
β2AR inhibition of pERK in primary HTMCs is mediated by Gαs.
To further study the ISO-mediated pERK increase in the presence of CTx pretreatment, various βAR ligands were tested for their ability to produce this effect in HTMCs (Fig. 4D). In these experiments, only the nonselective βAR agonist, cimaterol, and ISO produced statistically significant increases in pERK (15%±5%; and 11%±3% respectively). However, the endogenous ligand, epinephrine, as well as the β2AR selective agonists, procaterol and salbutamol, produced small nonsignificant increases in pERK. In contrast, timolol, ICI, and CGP had no effect at all on the pERK level of HTMCs.
β2AR-Gαs decrease in pERK is dependant on cAMP and PKA
Since the β2AR-pERK response in HTMCs is mediated by Gαs, it was next determined whether this effect was the result of the classic Gαs signaling pathway, activation of adenylyl cyclase, increased cAMP, and activation of PKA. To assess the roles of adenylyl cyclase (AC) and cAMP, the effect of forskolin, a direct activator of AC, on the pERK level in HTMCs was examined (Fig. 5A). In these experiments, forskolin treatment resulted in a statistically significant 15%±4% (P<0.01) and 20%±4% (P<0.001) decrease in pERK at 1 and 10 μM concentrations, respectively. These findings suggest that the ISO response can be mimicked by increasing the activity of adenylyl cyclase, which, in turn, increases the concentration of cAMP. In assessing the role of PKA in the forskolin-mediated decrease in pERK, the PKA inhibitor, H89 (5 μM), was found to significantly attenuate the foskolin response at 10 μM (P<0.05), thus suggesting that the forskolin response is dependant on PKA.
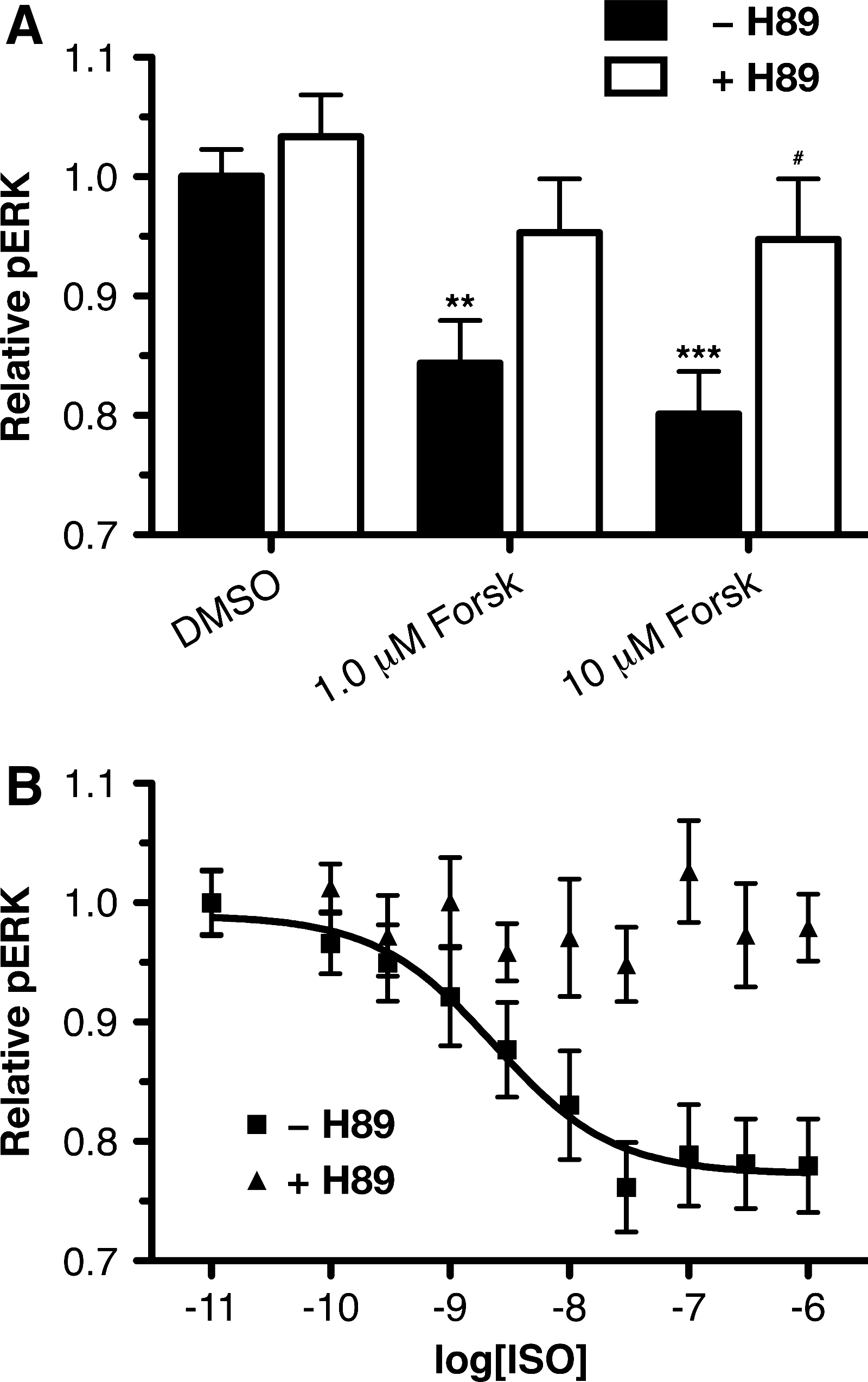
β2AR decrease in pERK in HTMCs is dependant on cAMP and PKA.
Having demonstrated that foskolin can mimic the β2AR-pERK response in HTMCs, and that the forskolin effect is attenuated by the PKA inhibitor, H89, a direct role for PKA in the β2AR-mediated pERK response was examined. ISO pERK dose-response curves were generated in the absence and presence of the PKA inhibitor H89 (Fig. 5B). In the absence of H89, ISO produced a dose-response curve with a maximum response of 22%±3% and pEC50 of 8.6±0.3. However, in the presence of H89 pretreatment, ISO treatment no longer produced a decrease in pERK, with a dose-response curve that did not converge. Statistically significant inhibitions of the ISO-pERK responses by 5.0 μM H89 were observed at ISO concentrations of 0.03 (P<0.01), 0.1 (P<0.001), 0.3 (P<0.001), and 1.0 μM (P<0.01). These findings imply that β2AR activation leads to a reduction in pERK through a mechanism that involves PKA, and likely adenylyl cyclase and cAMP.
The β2AR decrease in pERK is mediated by phosphatase activity
Since ISO treatment resulted in an immediate reduction in the level of pERK levels of HTMCs (data not shown), a potential role for phosphatases in this ISO-mediated effect was examined. ISO-pERK dose responses were obtained in HTMCs in the absence or presence of the nonselective protein tyrosine phosphatase inhibitor, vanadate (Fig. 6A). In these experiments, the presence of vanadate significantly inhibited the β2AR response at 10 (P<0.05), 30 (P<0.01), 100 (P<0.001), and 300 (P<0.01) nM concentrations of ISO. To confirm these findings, the ability of another nonselective protein tyrosine phosphatase inhibitor, phenylarsine oxide (PAO), was also examined in HTMCs (Fig. 6B). In the case of PAO, a significant reduction in the β2AR pERK response was observed at 30 (P<0.001), 100 (P<0.001), and 300 nM (P<0.001) concentrations of ISO.
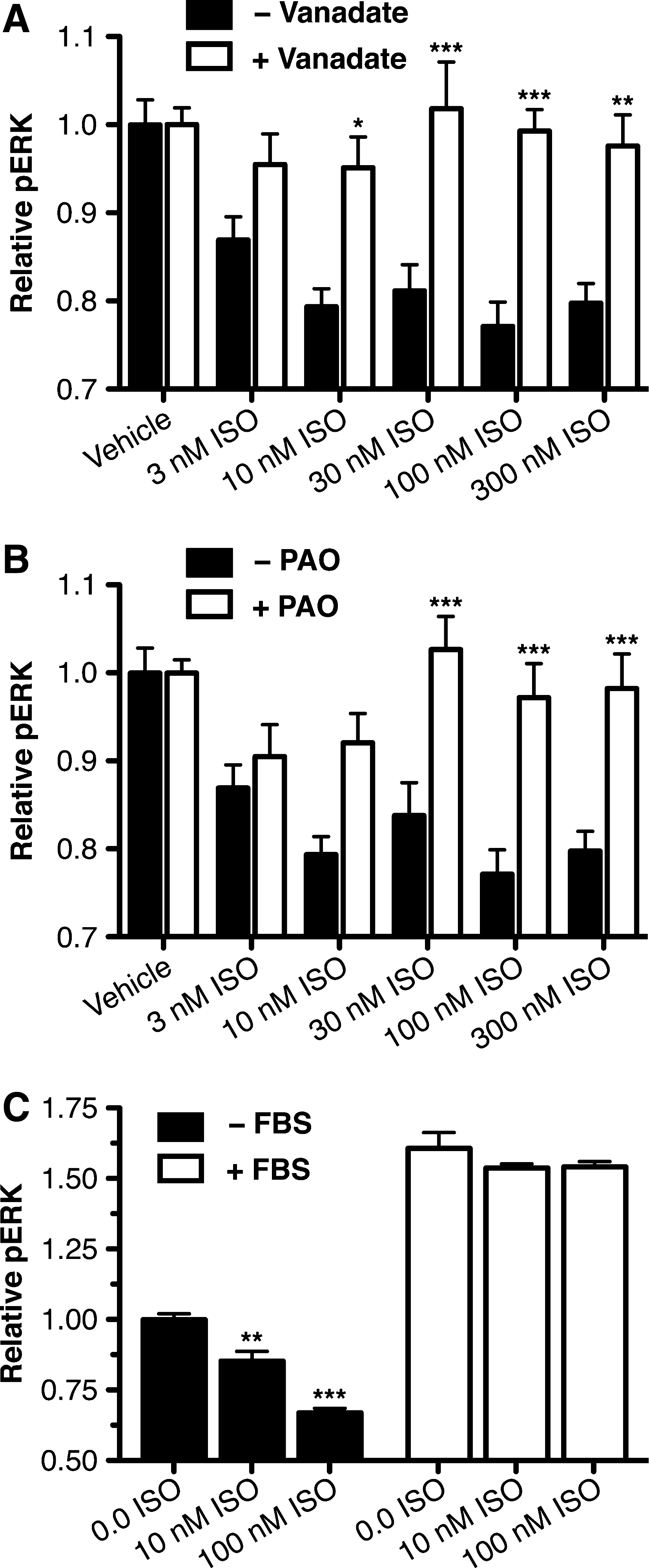
β2AR inhibition of pERK in HTMCs is dependant on phosphatase activity.
To further support these findings demonstrating that the ISO-pERK response is mediated by a phosphatase, the ability of ISO to inhibit pERK activation by FBS was considered (Fig. 6C). In this experiment, although ISO significantly reduced the basal pERK level of HTMCs after either 10 (P<0.01) or 100 nM (P<0.001) treatment, ISO did not significantly affect the strong FBS-pERK activation (61%±5%) at any concentration tested. This observation is consistent with ISO acting through a mechanism that is competitive with the activation of pERK by FBS. Collectively, these findings suggest that β2AR mediates a reduction in the pERK level of HTMCs through activation of a protein phosphatase.
Discussion
The TM is an important tissue in the regulation of IOP and represents a key target for the development of future IOP-lowering therapeutics for the treatment of glaucoma. 25 Although the IOP-lowering effect of βAR agonists on this tissue has been established,6,7 the specific cellular signaling pathways leading to this effect are still not clear. The current work demonstrates that in primary HTMCs, βAR agonists activate β2AR to activate competing pERK signaling pathways; one dependant on Gαs, AC, and cAMP, which reduced pERK, and one independent on both Gαs and Gαi/o, which increase pERK. In concert with previous reports that the IOP-lowering effects of βAR agonists in the TM correlate with increased cAMP, 6 and the observation that inhibition of ERK in these cells decreases ECM synthesis likely leading to increased AH outflow, 19 these findings indicate that the β2AR-pERK inhibitory pathway may play a critical role in the ability of βAR agonists to reduce IOP in vivo.
To date, the majority of previous studies on βAR pERK signaling have focused on the capacity of these receptors to increase in pERK in heterologous expression systems.10,26–28 This ability of βARs to activate pERK has been linked to many different signaling pathways including those involving Gαs, Gαi, G βγ, and β-arrestins.9–12 Given these previous findings, the fact that β2AR activation leads to a decrease in pERK in HTMCs is perhaps somewhat surprising. A decrease in pERK after β2AR activation has previously been reported in only 2 other cell types, specifically in keratinocyte and corneal epithelial cells.14–16 However, in these cells, unlike in HTMCs, the β2AR-pERK signaling pathway was not dependant on cAMP and PKA.14,16
It is also important to note that previous work examining the effect of cAMP signaling on pERK has demonstrated that increased cAMP often has an inhibitory effect on pERK.13,29 However, in these experiments, unlike our findings, increased cAMP did not decrease the basal pERK level, but instead inhibited pERK activation by various growth factors. In this case, the cAMP effect appears to be mediated by inhibition of the Raf-1 activation step in the ERK signaling cascade, 30 and not by phosphatase activity, as it was in the HTMCs. Based on this, and the fact that β2AR activation in HTMCs did not inhibit FBS-mediated pERK activation, the pERK signaling pathway reported here in HTMCs is unique from the previously described cAMP effects on pERK.
Another important finding of this work is the observation that after blocking the Gαs-meditated decrease in pERK in HTMCs, β2AR activation in these cells instead produced an increase in pERK. The fact that this β2AR increase in pERK was also not blocked by PTx suggests that this pathway is most likely through a β-arrestin mediated pathway. 11 In addition, there appears to be some degree of functional selectivity in comparing the Gαs inhibitory pathway with the non-Gαs/Gαi activating pathway of HTMCs; there are differing rank orders of efficacy for the βAR agonists tested in these 2 signaling pathways.31–33 Specifically, the rank order of efficacy for βAR agonists for decreasing pERK in HTMCs was ISO > cimaterol > procaterol > salbutamol > epinephrine; whereas the rank order of βAR agonists to increase pERK was cimaterol > ISO ≈ procaterol ≈ salbutamol ≈ epinephrine. These findings suggest that cimaterol signaling results in a conformation of β2AR that facilitates increased signaling through the pERK-activation pathway compared with the other ligands. 34 Such enhanced signaling has previously been demonstrated for several βAR ligands, thus biasing their signaling toward the β-arrestin pathway. 11
The presence of these 2 competing β2AR-pERK signaling pathways in HTMCs raises the question as to what cellular factors in HTMCs lead to the inhibitory pathway being predominant. Previous studies have demonstrated that the specific isoform of adenylyl cyclase expressed in HEK 293 cells determines the ability of cAMP to either activate or inhibit pERK. 35 In this study, it was found that the activation of the AC1 subtype in HEK 293 cells leads to an activation in pERK, whereas activation of the AC5 subtype appeared to produce a decrease in pERK. Alternatively, another study demonstrated that RNA silencing of the phosphodiesterase enzyme, PDE4D5, potentiated the ability of ISO to activate pERK in 293H cells expressing β2AR. 36 The findings of these 2 studies indicate that high expression levels of either AC5 or PDE4D5 could contribute to shifting the β2AR-pERK signaling pathway toward the inhibitory pathway; it will be important for future studies to examine the levels of these 2 enzymes in the HTMCs to determine whether they may individually play a role in determining the β2AR-pERK response in these cells.
Finally, it is also important to consider how these competing β2AR-pERK signaling pathways may influence the ability of βAR agonists to increase AH outflow, and reduce IOP. Previous reports have suggested that β2AR facilitates AH outflow through a pathway that is dependant on cAMP.6,7 Since β2AR-pERK inhibition in HTMCs is also dependant on cAMP, this suggests that it is the β2AR-pERK inhibition pathway, and not the competing β2AR-pERK activation pathway that may play a role in AH outflow. In support of this hypothesis, previous studies have shown that a decrease in pERK is associated with reduced cell migration and wound healing,14,16 a finding which suggests that a reduction in pERK by βAR agonists could produce the cytoskeletal changes in HTMCs that would be required in vivo to facilitate increased AH outflow, and the resulting reduction in IOP. 25 In support of this, a, a recent study demonstrated that the selective ERK inhibitor, U-0126, was able to inhibit ECM synthesis within the TM. 19 Such decreases in ECM are associated with increased AH outflow, 17 and indeed suggest that the inhibition of ERK by β2AR may directly facilitate a decrease in IOP in vivo.
The findings of the current work demonstrate that HTMCs possess competing β2AR signaling pathways leading to either a decrease or an increase in pERK. The fact that these 2 β2AR pERK pathways directly oppose each other suggests that they may be an ideal target for the identification of therapeutically useful functionally selective ligands. Functionally selective ligands are ligands that activate a specific signaling outcome more effectively than other signaling outcomes from the same receptors. 37 Recently, functionally selective ligands have been receiving increased attention for their ability to produce improved therapeutic efficacy, but reduced adverse side effects. 37 Specifically, in the case of β2AR signaling in the TM, if the β2AR-pERK decrease is responsible for the AH outflow effect of βAR agonists, then drugs that activate only this pathway, and not the competing pERK increasing pathway, may have a larger effect on outflow and, thus, generate a greater ocular hypotensive response. Further, such an approach may have the added benefit of identifying βAR agonists with improved side-effect profiles, a primary reason that βAR agonists have lost favor clinically in the treatment of glaucoma. 38 Future work will need to address whether such ligands for β2AR can be developed, and whether they may be indeed valuable as a novel, more effective means of targeting β2AR receptors for the control of IOP.
Footnotes
Acknowledgments
This work was supported by NSERC and Killam Trust studentships (B.D.H.) and a CIHR operating grant (M.E.M.K.).
Author Disclosure Statement
No competing financial interests exist.