
Abstract
Abstract
Müller glia, the principal macroglia of the retina, express diverse subtypes of adenosine and metabotropic purinergic (P2Y) receptors. Müller cells of several species, including man, also express ionotropic P2X7 receptors. ATP is liberated from Müller cells after activation of metabotropic glutamate receptors and during osmotic and mechanical induction of membrane stretch; adenosine is released through equilibrative nucleoside transporters. Müller cell-derived purines modulate the neuronal activity and have autocrine effects, for example, induction of glial calcium waves and regulation of the cellular volume. Glial calcium waves induced by neuron-derived ATP mediate functional hyperemia in the retina. Purinergic signaling contributes to the induction of Müller cell gliosis, for example, of cellular proliferation and downregulation of potassium channels, which are important for the homeostatic functions of Müller cells. Purinergic glial calcium waves may also promote the long-range propagation of gliosis and neuronal degeneration across the retinal tissue. The osmotic ATP release is inhibited under pathological conditions. Inhibition of the ATP release may result in osmotic Müller cell swelling and dysregulation of the water transport through the cells; both may contribute to the development of retinal edema. Suppression of the osmotic ATP release and upregulation of the ecto-apyrase (NTPDase1), which facilitate the extracellular degradation of ATP and the formation of adenosine, may protect neurons and photoreceptors from death due to overactivation of P2X receptors. Pharmacological inhibition of P2X7 receptors and stimulation of adenosine receptors may represent clinical approaches to prevent retinal cell death and dysregulated cell proliferation, and to treat retinal edema.
Introduction
T

Cellular constituents of the retina. Müller cells span the entire thickness of the neuroretina. The perikarya of Müller cells are localized in the INL. The funnel-shaped endfeet of Müller cells form the inner surface of the retina. Both AG and Müller cells contact the superficial blood vessels, nerve fibers, and the inner surface of the retina. MG are located in both plexiform layers and the GCL. A, amacrine cell; AG, astrocytes; B, bipolar cell; C, cone; G, ganglion cell; GCL, ganglion cell layer; H, horizontal cell; INL, inner nuclear layer; IPL, inner plexiform layer; M, Müller cells; MG, microglia; ONL, outer nuclear layer; OPL, outer plexiform layer; PRS, photoreceptor segments; R, rod; RPE, retinal pigment epithelium.
Müller cells are the anatomical and functional link between retinal neurons and compartments outside the neuroretina (blood vessels, vitreous chamber, subretinal space). Müller cells have manifold functions that support the neuronal activity, including the delivery of nutrients to neurons, the removal of “waste” products, the mediation of extracellular potassium and water homeostasis, and the regulation of the extracellular space volume and retinal blood flow. 1 Müller cells release “gliotransmitters,” which modify the neuronal activity, and are crucially involved in the maintenance of the synaptic activity in the inner retina by neurotransmitter recycling and the delivery of neurotransmitter precursors. 1 Müller cells are living optical fibers that transfer the light to the photoreceptors. 2 In addition, Müller cells buffer mechanical tissue deformations. 2
Müller cells are activated by pathogenic stimuli. Müller cell gliosis is characterized by a “Janus face.” Early after tissue injury, gliosis is neuroprotective and represents an attempt to limit the severity of injury. However, reactive Müller cells may also quit neuronal support, contribute to neurodegeneration, and impede regular regeneration of the retinal tissue. 3
Activation of purinergic receptors by purines such as ATP and adenosine contributes to the regulation of the neuron-supportive functions of Müller cells. 4 Purines such as ATP and adenosine are neuro- and gliotransmitters in the retina, and are involved in transmission of signals between neurons and glial cells.4–8 Photoreceptors, neurons, glial cells, the vasculature, and the retinal pigment epithelium express P1 (adenosine) and P2 (nucleotide) receptors. 6 Under pathophysiological conditions, purinergic signaling is not only involved in inducing retinal gliosis and degeneration of photoreceptors, neurons, and vascular cells but may also protect the retinal tissue from degeneration.4,8
All pathological conditions investigated so far are associated with activation of purinergic receptor signaling in the retina. Activation of purinergic receptors is often crucially involved in the induction of signal transduction and effector cascades that are implicated in mediating degenerative alterations of the retina. In this review, we give a short overview of the present knowledge regarding the functional impact of purinergic receptor signaling in Müller cells under normal and pathological conditions.
Purinergic Receptors of Müller Glia
Müller cells of all vertebrate species investigated so far express various subtypes of metabotropic purinergic (P2Y) receptors, including P2Y1 and P2Y4 (Fig. 2A, B).9–11 Different P2Y receptor subtypes are unevenly distributed in distinct Müller cell membrane domains (Fig. 2A, B). The predominant receptor subtypes that induce calcium responses in rodent Müller cells are P2Y1 (Fig. 3D), and in rabbit Müller cells P2Y1 and P2Y4 (Fig. 3C).12–15 Activation of P2Y receptors in human Müller cells elicits 3 responses: a transient release of calcium from intracellular stores, a transient activation of calcium-activated cation channels, and a more sustained influx of calcium from the extracellular space (Fig. 4A–C). 16
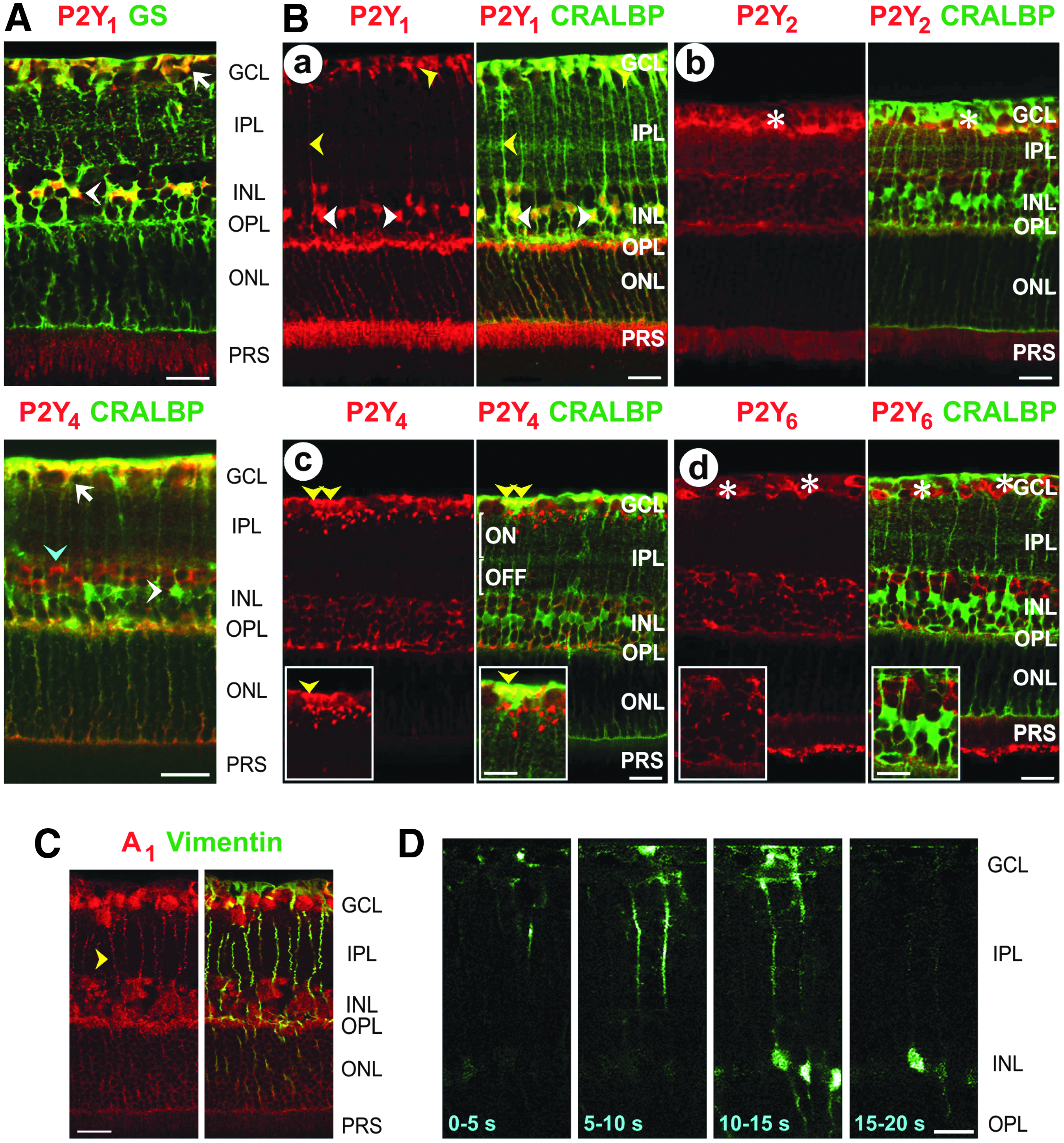
Purinergic receptors of rodent Müller cells. Retinal slices were costained against the glial cell markers GS, CRALBP, and vimentin, respectively; colabeling yielded a yellow-orange merge signal.

Purinergic calcium responses in Müller cells.
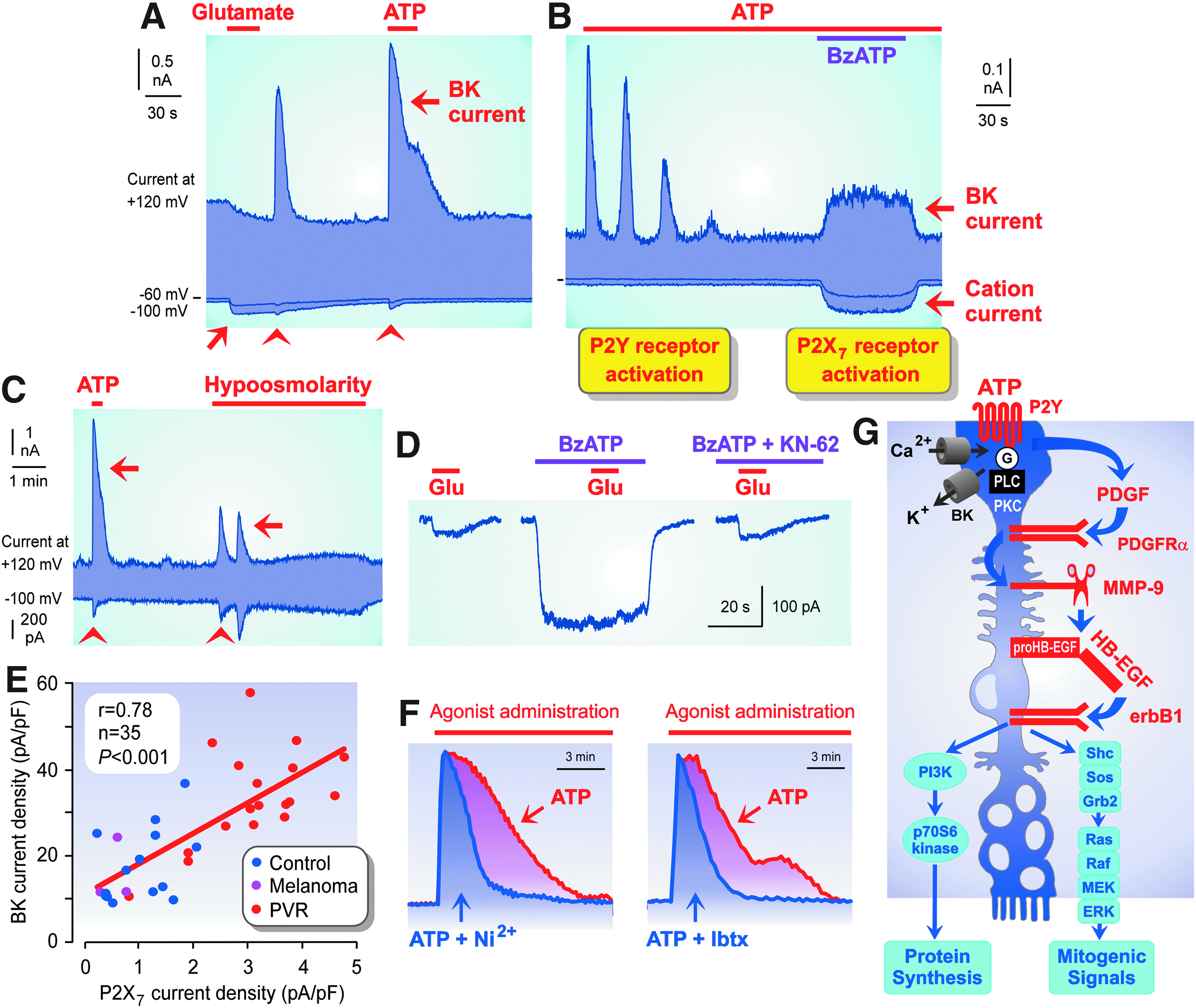
P2Y and P2X7 receptor signaling in human Müller cells and P2Y receptor-induced Müller cell proliferation.
Ionotropic purinergic (P2X) receptors are ATP-gated cation channels. The expression of P2X receptor subtypes in Müller cells is species dependent. Human, monkey, and chick Müller cells, and cells of lower vertebrates, express P2X7 receptors.17–20 Activation of P2X7 receptors in human Müller cells by ATP or benzoylbenzoyl-ATP (BzATP) induces plasma membrane depolarization, an influx of calcium through P2X7 receptor channels, and opening of calcium-activated, big-conductance potassium (BK) channels (Fig. 4B).16,17,21 Rat Müller cells contain gene transcripts of P2X3,4,5 receptors and display P2X4 immunoreactivity22,23; however, stimulation with purinergic receptor agonists does not alter the membrane conductance of the cells, which would suggest the presence of functional P2X receptors.21,24
Müller cells also express various adenosine receptor subtypes, including A1 (Fig. 2C), A2A, and A2B receptors. 25 In rat Müller cells, activation of adenosine receptors induces calcium responses or potentiates light-induced calcium responses.13,25 Adenosine does not induce a calcium response in Müller cells of various other species such as rabbit (Fig. 3C) and man.4,15,16 Activation of adenosine receptors also triggers calcium-independent responses implicated, for example, in the homeostasis of the Müller cell volume (see section Impairment of Homeostatic Müller Cell Functions).
Release of Purines from Müller Glia
In the retina, purines are preferentially released in the dark and during neuronal activity.26,27 By activation of P2X receptors, synaptically released ATP contributes to the fast excitatory neurotransmission. P2X receptors are expressed by most types of retinal neurons.6,7 Extracellular ATP also modulates the neuronal activity by activation of P2Y receptors on neuronal and glial cells.4,6 Adenosine inhibits the neuronal activity via various mechanisms, including suppression of the neurotransmitter release. 6 Adenosine is released from neuronal and glial cells through equilibrative nucleoside transporters; in various species and under pathological conditions, adenosine can be also generated in the extracellular space by the enzymatic degradation of ATP.28–31
Various receptor-dependent and receptor-independent stimuli induce a release of ATP from Müller cells. Osmotic (Fig. 4C) and mechanical stress, as well as electrical stimulation, induces a release of ATP from Müller cells; in addition, ATP is liberated after activation of purinergic, thrombin, dopaminergic, and glutamatergic receptors (Figs. 4A and 5C).14,28,30–34 In dependence on the species investigated, the glutamate-induced ATP release from rodent Müller cells depends on intracellular calcium signaling. The glutamate-induced release of ATP from rat Müller cells is mediated by a calcium-independent mechanism,30,31 while liberation of calcium from inositol triphosphate (IP3)-gated internal stores is a precondition for the release of ATP from murine Müller cells. 14

Alterations in the glial extracellular nucleotide metabolism and Müller cell swelling characteristics in experimental diabetic retinopathy in the rat.
Adenosine is released from rodent Müller cells through calcium-independent equilibrative nucleoside transporters.30,31 In the retinal parenchyma of mice, adenosine can be also formed from ATP in the extracellular space. 14 The consecutive dephosphorylation of ATP is mediated by the ecto-ATPase (nucleoside triphosphate diphosphohydrolase-2, NTPDase2), ecto-apyrase (NTPDase1), and ecto-5′-nucleotidase (CD73). The parenchyma of the rat retina does not contain NTPDase1, which generates AMP, the substrate of the ecto-5′-nucleotidase (Fig. 5A).35,36 However, in the retina of diabetic rats, the expression of NTPDase1 is upregulated (Fig. 5A); this makes it possible that adenosine is extracellularly generated from ATP (Fig. 5C). 36
Autocrine/paracrine ATP signaling mediates the propagation of long-range glial calcium waves (Figs. 3D, E, and 6D).12,37 The calcium responses are transmitted from astrocytes to Müller cells, and between Müller cells, by a release of ATP and a P2Y1 receptor-mediated release of calcium from intracellular stores.12,32,33 The calcium waves can be triggered by light (Fig. 6D), electrical and mechanical stimuli, and transmitters such as glutamate.12,37,38 Calcium waves may transmit glial signals across larger areas of the retina, to synchronize the activities of a multitude of neurons that form a functional unit. Glial calcium waves also mediate the neurovascular coupling and may transmit cell volume-regulatory signals (see section Regulation of Retinal Blood Supply). After a focal retinal injury, glial calcium waves may contribute to the spread of retinal gliosis and degeneration to larger retinal areas (see section Spatial Propagation of Müller Cell Gliosis).

Neuronal activity, induced by repetitive light stimulation of the guinea-pig retina, is sensed by Müller cells via inducing glial calcium responses.
Modulation of Neuronal Activity
Glial calcium waves are accompanied by alterations in the activities of neurons. When the waves reach the ganglion cell layer neurons, the light-induced spike rate is decreased in ∼50% of the neurons; further neurons display excitation. 37 The inhibition of the light-induced activity was suggested to result from activation of neuronal A1 receptors by adenosine, which is extracellularly generated from Müller cell-derived ATP. 28 ATP released from Müller cells may induce neuronal activation by stimulation of P2X receptors. Both glia- and neuron-derived ATP 39 may contribute to the overstimulation and degeneration of photoreceptors, retinal ganglion cells, and amacrine cells under pathological conditions such as hypoxia, glaucoma, and inherited photoreceptor degeneration.40–44 Since mechanical stimuli induce a release of ATP from Müller cells,28,32,33 excess ATP may be liberated from Müller cells under pathological conditions such as high intraocular pressure and retinal detachment.40,45 ATP released from perivascular Müller cell processes may contribute to the degeneration of the retinal microvasculature in diabetic retinopathy, which has been suggested to be mediated, at least in part, by a P2X7-mediated lethal calcium influx. 46 Adenosine released from Müller cells has been also suggested to contribute to the protection of photoreceptors during hypoxic states in the dark. 29 The extracellular level of adenosine is increased in the dark; the increase results from inhibition of the cellular uptake of adenosine and a stimulated degradation of extracellular ATP liberated by neurons and Müller cells. 29
Light-Induced Glial Calcium Responses
In tissue preparations of the guinea-pig retina, light stimulation induces 2 different calcium responses in Müller cells: the immediate response is induced in the whole Müller cell bodies and develops slowly, while delayed calcium responses appear with a latency of ∼3 min and had transient kinetics (Fig. 6A–C). 38 The immediate response is a calcium influx from the extracellular space triggered by zinc ions released from photoreceptor cells 47 and by hyperpolarization of the plasma membrane; the latter is induced by the decreased activity of photoreceptors, which results in a decrease of the extracellular potassium concentration and the lowered activation of electrogenic glutamate transporters (Fig. 6E, F). 38 The transient responses are triggered in the Müller cell endfeet and propagate through the Müller cell bodies toward the layers of photoreceptor cell bodies and synapses (Fig. 6D). 38 These responses are mediated by a release of calcium from internal stores induced, in part, by ATP released from amacrine and ganglion cells25,27 and from Müller cells (in response to osmotic stress; see section Impairment of Homeostatic Müller Cell Functions) (Fig. 6G). 38
The immediate calcium response may support the neuronal signal transmission from photoreceptors to ganglion cells; the hyperpolarization increases the efficiency of the glial electrogenic glutamate uptake, and the calcium response may induce activation of calcium-dependent potassium channels (which increases the glial potassium buffering capacity) and may also trigger a calcium-dependent release of glutamate, which inhibits an osmotic swelling of Müller cells (see section Impairment of Homeostatic Müller Cell Functions). The delayed calcium responses may reflect a feedback signaling from ganglion cells to photoreceptors through Müller cells. 38 This signaling may regulate the activity of photoreceptors in dependence on the activity of retinal ganglion cells; overactivation of ganglion cells may inhibit the activity of photoreceptor synapses via the release of adenosine by Müller cells.
Regulation of Retinal Blood Supply
Because the retina has extremely high glucose and oxygen demands, 48 regulation of the blood flow is crucial to support the retinal function. 49 Light triggers a dilation of retinal arterioles, which are localized at the inner retinal surface, a phenomenon called functional hyperemia. 49 Light stimulation of retinal tissue preparations induces a release of ATP from retinal neurons in the inner plexiform layer; ATP triggers calcium responses in Müller cells (Fig. 6A, G).38,50 The calcium signals are transmitted to the retinal surface via propagation of ATP-mediated glial calcium waves.25,50 Calcium-dependent phospholipases A2 produce arachidonic acid; metabolites of arachidonic acid cause dilation (epoxyeicosatrienoic acids and prostaglandins) or constriction (20-hydroxyeicosatetraenoic acid) of arterioles.50,51 Whether vasodilation or constriction is induced depends on the production of nitric oxide, which inhibits vasodilation.50,52 ATP released from perivascular glial processes may also directly constrict retinal arterioles (Fig. 3E). 53 Neuron- or glia-derived ATP and adenosine also induce constriction and relaxation, respectively, of capillary-surrounding contractile pericytes.54–56
Diabetic retinopathy is associated with a loss of functional hyperemia that leads to retinal hypoxia. 57 In experimental diabetic retinopathy, the glia-induced vasodilation is suppressed likely because of the upregulation of the inducible nitric oxide synthase. 52 Because ATP-induced glial calcium waves contribute to the mediation of functional hyperemia, 51 it is conceivable that the alteration of the glial calcium responsiveness under pathological conditions (see section Induction of Müller Cell Gliosis) may represent 1 mechanism, which underlies the impairment of the neurovascular coupling in diabetic retinopathy.
Induction of Müller Cell Gliosis
Extracellular ATP is a danger signal that is released from cells under pathological conditions such as inflammation, mechanical, osmotic, and oxidative stress, hyperglycemia, increase of intraocular pressure, and ischemia/hypoxia.40,58,59 Under various pathological conditions, Müller cells display an increase of the calcium responses induced by activation of purinergic receptors. The incidence of Müller cells, which show calcium responses after activation of P2Y receptors, is dependent on the species investigated. In the retinas of rats, guinea pigs, and man, the vast majority of Müller cells show a calcium response on activation of P2Y receptors by ATP (Fig. 3A, D),13,16 while in the retinas of rabbits and pigs, only few Müller cells (∼10% of all cells) respond to ATP with an increase of the cytosolic-free calcium level (Figs. 7 and 8E).15,60,61 The incidence of Müller cells, which display ATP-induced increases of the cytosolic-free calcium level, is strongly increased in the retinas of rabbits and pigs under various pathological conditions such as retinal ischemia, detachment, and proliferative vitreoretinopathy (PVR) (Figs. 7 and 8E, F).15,60–63 Müller cells of patients with PVR show increased calcium-activated cation currents, which are activated on stimulation of P2Y receptors (Fig. 4A, C) and increased cation currents through P2X7 receptor channels (Fig. 4E).16,21 The latter suggests an increased expression of P2X7 receptors in Müller cells.

Upregulation of purinergic calcium responsiveness in Müller cell gliosis. The incidence of rabbit Müller cells, which display ATP (200 μM)-induced calcium responses (diagram, below), is inversely related to the density of inwardly rectifying potassium (Kir) currents (diagram, above). This relationship was observed during the postnatal development (left side) and during experimental retinal detachment and moderate and massive PVR (right side). The traces above represent examples of whole-cell potassium current records in single isolated cells. Below: Examples of peak calcium responses (green areas) to ATP (200 μM) at the inner surface of rabbit retinas. While nearly all Müller cell endfeet display a calcium response in the retina of the postnatal day-8 rabbit and of rabbits with retinal detachment and PVR, only some Müller cell endfeet responded in the retina of the adult control animal. Yellow and red colors are light reflections at nerve fibers and microglial cells. Modified from Uhlmann et al. 113

Propagation of retinal gliosis in response to experimental focal detachment of the porcine retina. The tissues and cells were obtained from control retinas, from retinal areas that were detached for 3 and 7 days, respectively, and from nondetached retinal tissues that were localized near or distant to the focally detached retinal area in situ (peridetachment).
The increase of the calcium responses, which are triggered by activation of purinergic receptors, is inversely related to the amplitude of potassium currents mediated by inwardly rectifying potassium (Kir) channels (Figs. 7 and 8C, D).15,21,61 The expression of Kir channels is an event occurring during the differentiation of Müller cells from progenitor cells in the developing retina. 64 Therefore, the reduction of the Kir currents and the increase of the calcium responses, which are triggered by activation of purinergic receptors, may reflect a dedifferentiation of Müller cells under pathological conditions; both alterations are related to the strength of gliosis (Fig. 7).
It was suggested that activation of purinergic receptors is a critical step in the initiation of Müller cell gliosis and may trigger the upregulation of intermediate filaments (Fig. 8A), the hypertrophy (Fig. 8B) and proliferation of Müller cells (see section Proliferation of Müller Cells), and the downregulation of functional Kir channels (Figs. 7 and 8C, D).15,61,65,66 The reduction of the Kir currents observed in gliotic Müller cells is, in part, inhibited in the presence of suramin (a blocker of nucleotide and growth factor action) and in P2Y1-knockout mice.15,67 In a rat model of chronically increased intraocular pressure, the downregulation of Kir channels in Müller cells was attributed to an overactivation of metabotropic glutamate receptors. 68 It has been shown that stimulation of metabotropic glutamate receptors in Müller cells triggers a release of ATP (Figs. 4A and 5C). 30 Therefore, it may be possible that glutamate induces a transactivation of P2Y1 receptors in Müller cells resulting in downregulation of Kir channels. However, the purinergic contribution to the induction of gliotic alterations of Müller cells remains to be elucidated in more detail.
Impairment of Homeostatic Müller Cell Functions
Gliosis is associated with an impairment of the homeostatic functions of Müller cells; impaired tissue homeostasis contributes to the secondary tissue damage after a primary retinal injury. Potassium currents through Kir channels are the major determinant of the very negative membrane potential, which is a precondition of various voltage-dependent functions of Müller cells. 1 The downregulation of Kir4.1 channels (Figs. 7 and 8C, D), which is mediated (at least in part) by purinergic signaling (see section Induction of Müller Cell Gliosis), results in membrane depolarization that impairs the regulation of the local extracellular potassium level and the neurotransmitter uptake through electrogenic transporters. 1 Chick and human Müller cells also depolarize during activation of P2X7 receptors; depolarization impairs the glutamate uptake (Fig. 4D), 17 which results in extracellular accumulation of glutamate and excitotoxic death of neurons. 19
Extracellular ATP induces a release of growth factors such as the basic fibroblast growth factor (bFGF) from Müller cells. 69 bFGF induces a downregulation of the glutamine synthetase in Müller cells. 70 Although bFGF is known to support the survival of photoreceptors and neurons, 3 the ATP/bFGF-induced decrease of glutamine synthetase expression may contribute to neuronal degeneration.
Glutamatergic neurotransmission triggers an influx of sodium chloride and water into neurons; the ion influx results in a decrease of the extracellular osmolarity, 71 and the water influx causes a swelling of neuronal cell structures such as somata and synapses (Fig. 5B).31,72,73 Müller cells control the volume of the extracellular space; they adjust their morphology to the varying size of neurons and inhibit the swelling of their cell bodies and of bipolar cells under hypoosmotic conditions.69,72,74 These alterations prevent adverse decreases of the extracellular space volume, which induces neuronal hyperexcitation. A swelling of Müller cells under hypoosmotic conditions is prevented by at least 2 mechanisms: by Kir4.1-mediated potassium currents and by autocrine stimulation of a purinergic receptor signaling pathway.30,75
The purinergic pathway is mediated by ATP and adenosine released from Müller cells, which activate P2Y1 and A1 receptors (Fig. 5C). Stimulation of A1 receptors activates potassium and chloride channels; the efflux of potassium and chloride ions restores the osmotic balance across the cell membrane. In addition to extracellular hypoosmolarity, the purinergic pathway is activated by glutamate released from neurons or Müller cells (Fig. 5C).31,72 The prevention of the osmotic swelling of bipolar cells, which lack an autocrine volume regulation, is also initiated by the glial purinergic receptor signaling (Fig. 5C).69,74
Müller cells lose their capability of cell volume regulation under pathological conditions such as retinal inflammation, detachment (Fig. 8G), ischemia–reperfusion, diabetic retinopathy (Fig. 5B), and retinal degeneration induced by excess blue light.31,36,61,63,75–79 The impairment of cell volume regulation was attributed to both the downregulation of Kir4.1 channels (Figs. 7 and 8C, D) and a suppression of the ATP release induced by osmotic-mechanical stimuli.4,80 The extent of Müller cell swelling observed under hypoosmotic conditions is inversely related to the amplitude of the Kir currents (Fig. 8H).63,78 The suppression of the ATP release from Müller cells may serve for self-protection, that is, it prevents overactivation of purinergic receptors and thus cytotoxic calcium rises. On the contrary, the swelling of Müller cells, which may occur in osmotic stress, results in decreased volume of the extracellular space; such volume decreases may contribute to overactivation and degeneration of retinal neurons.
The osmotic Müller cell swelling is a sign of a dysregulation of the transmembrane water transport; such dysregulation may contribute to the development of retinal edema by inducing a water accumulation within Müller cells and by a disruption of the water clearance from the retinal tissue, which occurs through Müller cells (Fig. 9A). 80 A water accumulation within Müller cells was observed in animal models of retinal ischemia/hypoxia, diabetic retinopathy, retinal detachment (Fig. 9B), and retinal light injury,81–86 and in the human retina after light injury. 87

Müller cell gliosis contributes to the secondary retinal degeneration after a primary insult.
Unlike the ATP release induced by osmotic stimuli, the release of ATP triggered by glutamate (which does not induce calcium responses in Müller cells of rats and guinea pigs; Fig. 3A, B)12,25,30,72 continues to be active under pathological conditions.4,30 Various bioactive signaling molecules were shown to trigger a release of glutamate from Müller cells, which impedes the osmotic swelling via activation of the purinergic receptor signaling pathway; these molecules include, for example, nerve growth factor, osteopontin, erythropoietin, neuropeptide Y, and vascular endothelial growth factor (Fig. 5B, C).30,31,34,69,88 Inhibition of Müller cell swelling may represent 1 mechanism of the neuroprotective action of these molecules.
Purinergic signaling in Müller cells is also involved in mediating the inflammatory cell death in the diabetic retina. Hyperglycemia induces autocrine purinergic receptor signaling in Müller cells via inducing a release of ATP and adenosine; activation of purinergic receptors stimulates the activity of caspase-1 and the secretion of interleukin-1β. 89 Interleukin-1β contributes to the degeneration of the diabetic retina.90,91
P2Y receptor signaling in Müller cells may also increase the survival of photoreceptors. In the murine retina, P2Y1 receptors are localized to Müller cells, retinal ganglion cells, and a subpopulation of amacrine cells.14,92,93 After retinal ischemia–reperfusion in P2Y1-knockout mice, the survival of amacrine cells is increased and the degeneration of photoreceptor cells is decreased compared to wild-type control. 67 These data support the assumption that P2Y1 receptor signaling in Müller cells improves the survival of photoreceptors, while activation of P2Y1 receptors in amacrine cells contributes to cell death under ischemic conditions. 67
Spatial Propagation of Müller Cell Gliosis
After focal injury of the retina such as retinal detachment, the ATP-mediated propagation of glial calcium waves (Fig. 2D) may contribute to a spread of Müller cell gliosis from the site of injury into the neighboring nondamaged tissue. Focal retinal detachment causes a time-dependent spatial propagation of Müller cell gliosis across the retina as indicated, for example, by the increased expression of intermediate filaments (Fig. 8A), Müller cell hypertrophy (Fig. 8B), the downregulation of functional Kir channels (Fig. 8C, D), the increased potency of ATP to induce cytosolic calcium rises in Müller cells (Fig. 8E, F), and the alteration in the osmotic swelling properties of Müller cells (Fig. 8G).15,61 The spatial propagation of Müller cell gliosis is accompanied by the induction of retinal degeneration, as indicated by the photoreceptor degeneration and the presence of edematous Müller cells and cysts (Fig. 9A, B). 86 It has been suggested that the retinal degeneration in the tissue not affected by the primary injury is caused by the disruption of the homeostatic Müller cell functions, which results in disturbed potassium and water homeostasis and impaired glutamate clearance. 94
In response to mechanical stress as occurring after retinal detachment, Müller cells release ATP and growth factors such as bFGF.28,32,33,95 It has been shown that there is a positive relationship between the actions of ATP and growth factors in Müller cells; ATP triggers the release of growth factors, and growth factors increase the sensitivity of P2Y receptors in Müller cells (see section Proliferation of Müller Cells). This bidirectional loop of ATP and growth factor action may provoke the secretion of soluble factors, which diffuse from the site of injury to noninjured retinal areas; extracellular ATP and growth factors trigger gliosis in retinal areas distant from the site of primary injury (Fig. 9C). 94
Proliferation of Müller Cells
Overstimulated proliferation of Müller cells is a characteristic of proliferative retinopathies such as PVR, which often evolves from retinal detachment. 3 Proliferating Müller cells display alterations in the potassium channel expression; whereas Kir channels are downregulated (reflected by the decrease of the whole cell inward potassium currents; Fig. 7), calcium-activated BK channels are upregulated.64,96 The downregulation of Kir channels causes a depolarization of the cells; therefore, the membrane potential can rapidly shift between depolarized and hyperpolarized values when voltage-gated sodium and calcium channels, P2X7 receptor channels, BK channels, and other potassium channels open and close. 64
Stimulation of P2Y and P2X7 receptors induces proliferation of Müller cells; to promote Müller cell proliferation, a calcium influx through voltage-gated calcium and/or P2X7 receptor channels is required.21,64,66,97 Activation of BK channels induces cellular hyperpolarization, which stimulates calcium currents through open P2X7 receptor channels (Fig. 4B); closure of BK channels induces membrane depolarization, which stimulates the activity of voltage-gated calcium channels. 64 The P2Y receptor-induced influx of calcium through voltage-gated calcium channels is increased by the activity of BK channels (Fig. 4F); the proliferation rate of Müller cells increases with the duration of the calcium responses. 66
The stimulatory effect of ATP on Müller cell proliferation is mediated by the action of growth factors. Activation of purinergic receptors induces a release of at least 2 growth factors from Müller cells, platelet-derived growth factor (PDGF) and heparin-binding epidermal growth factor-like growth factor (HB-EGF); activation of the epidermal growth factor (EGF) receptor tyrosine kinase by HB-EGF triggers the final steps of mitogenic signaling (Fig. 4G). 97 As a positive feedback regulation, HB-EGF also stimulates the release of ATP from Müller cells. 98 After activation by ATP, P2Y receptors are desensitized for a long time period. 99 Growth factors such as nerve growth factor, PDGF, and EGF rapidly induce a resensitization of P2Y receptors, which restores the ATP-induced calcium responses in Müller cells. 99
Adenosine-Mediated Retinal Protection
In the retina, adenosine receptors are expressed by neuronal and glial cells (Fig. 2C). 6 By suppression of excitatory neurotransmission, adenosine protects against glutamate-induced degeneration of retinal neurons. 6 Activation of adenosine receptors also prevents the P2X7 receptor-mediated apoptosis of retinal ganglion cells.42,100 The upregulation of NTPDase1 in Müller cells of the diabetic retina (Fig. 5A) 36 supports the survival of retinal neurons because it facilitates the degradation of neurotoxic ATP and increases the extracellular adenosine level. A higher rate of ATP/ADP degradation in the extracellular space resulting from upregulation of glial NTPDase1 may also support the survival of Müller cells because it prevents a cytotoxic calcium overload due to excess activation of P2Y1 receptors. In addition, the reduced retinal expression of adenosine deaminase and adenosine kinase contributes to the increase of the extracellular level of adenosine in the diabetic retina 101 (but see Ref. 59 ). In Müller cells, adenosine not only induces expression of Kir4.1 channels, 102 which supports the potassium homeostasis of the retina, but also a downregulation of glutamate transporters and glutamine synthetase, 103 which favors glutamate-induced neuronal cell death.
Retinal Edema
Müller cells control the osmotic balance within the retinal tissue and between blood and the retina by facilitating transmembrane water fluxes. Glial water fluxes are driven and directed by the currents of osmolytes, in particular of potassium ions. In the endfoot and perivascular membranes of Müller cells, aquaporin-4 water channels and Kir4.1 potassium channels are colocalized.80,104 Under various pathological conditions, the perivascular expression of Kir4.1 is reduced (Fig. 8C), whereas the expression of aquaporin-4 is unchanged or even increased. 84 The downregulation of Kir4.1 results in a dissociation of the water fluxes and potassium currents and favors an accumulation of potassium ions within Müller cells; both support an accumulation of water in the retinal tissue and within Müller cells (Fig. 9A, B). 80 The inhibition of the ATP release from Müller cells in response to osmotic stimuli, which may result in Müller cell swelling (Fig. 5B), contributes to the development of a cytotoxic retinal edema.
Anti-inflammatory corticosteroids such as triamcinolone acetonide are clinically used for the resolution of retinal edema. 105 Triamcinolone was shown to inhibit the osmotic swelling of Müller cells in animal models of various retinal disorders such as retinal ischemia/hypoxia, inflammation, and diabetic retinopathy.36,78,79,106 Triamcinolone stimulates the release of adenosine from Müller cells which, via activation of A1 receptors, triggers activation of potassium and chloride channels (Fig. 5C).31,106 In Müller cells of the diabetic retina, the swelling-inhibitory effect of triamcinolone is also mediated by the extracellular degradation of ATP through the action of NTPDase1 (Fig. 5C); this fact may suggest that it also triggers a release of ATP from Müller cells. 36 The activation of potassium and chloride channels may also support the release of osmolytes from perivascular Müller cell processes into the blood and the transcellular water transport through Müller cells; both may facilitate the clearance of excess water from the retina. 106 Promotion of the transglial water transport may represent a further mechanism of the neuroprotective effect of adenosine in the retina.
Conclusions
Currently, the many roles of purinergic signaling implicated in Müller cell (dys)function are still not resolved. Much knowledge was obtained in animal and culture models and must be confirmed in human tissues and subjects. There is evidence that gliotic alterations of Müller cells are an important cause of secondary neuronal degeneration after a primary retinal injury. These alterations impair various neuron-supporting functions of Müller cells, including the glutamate uptake, the regulation of the retinal potassium and water homeostasis, and the control of the extracellular space volume. Purinergic receptor signaling induces many aspects of physiological and degenerative retinal alterations under pathological conditions, and purinergic receptors represent potential targets for the pharmacological treatment of blinding retinal disorders. However, the development of pharmacological strategies is hampered by the fact that activation of 1 receptor subtype often has both beneficial and detrimental effects for the protection of different retinal cell types and for the preservation of different functional aspects, respectively. One example is the P2Y1 receptor; activation of P2Y1 receptors is implicated in the induction and spread of Müller cell gliosis and has contrary effects on the survival of photoreceptor and amacrine cells in the retina after transient ischemia. 67
Inhibition of P2X7 receptors, for example, by brilliant blue G, which is an adjuvant in retinal surgery, 107 may not only reduce the degeneration of retinal neurons and microvasculature in retinal ischemia/hypoxia, glaucoma, and diabetic retinopathy42–44,46,100,108 but may also protect from the impairment of glial voltage-dependent functions such as the uptake of glutamate (Fig. 4D),17,19 and may inhibit the uncontrolled proliferation of Müller cells in PVR. 21 However, the long-term therapeutic utility of P2X7 receptor antagonists is presently not proved and it is unclear whether P2X7 inhibition is efficient to inhibit cell death in the human retina.
Further pharmacological approaches to inhibit retinal cell death include the use of adenosine receptor agonists and adenosine kinase inhibitors. 109 It has long been known that pharmacological activation of adenosine receptors has protective effects against ischemic retinal injury. 110 Agonists of adenosine A1 receptors may also support the treatment of retinal edema. However, because adenosine may also induce a downregulation of transporters and enzymes implicated in retinal glutamate recycling, 103 a potential long-term beneficial effect of adenosine receptor agonists has to be proven in future experiments. Systemic side effects of the agents may be limited by the use of eye drops. It has been shown that P2Y receptor signaling stimulates the proliferation of retinal progenitor cells. 111 It is conceivable that agonists of purinergic receptors may be helpful to promote the proliferation and dedifferentiation of Müller cells in vitro; dedifferentiated Müller cells may be used as “stem cells” for retinal regeneration. 3
Footnotes
Acknowledgment
Some of the work presented in this review was supported by grants from the Deutsche Forschungsgemeinschaft (GRK 1097/1, RE 849/16-1).
Author Disclosure Statement
No competing financial interests exist.