
Abstract
Abstract
Necrobiotic xanthogranuloma (NXG) is a chronic, progressive non-Langerhans histiocytic granulomatous disease. While case reports describing periorbital involvement of NXG are frequent, only a few case reports describing ocular involvement, such as scleritis and uveitis, exist. Herein, we present a case presenting initially as bilateral anterior and posterior scleritis, as well as a chronic bilateral granulomatous panuveitis, and discuss the immunosuppressant options that should be considered for this disease with protean manifestations.
Introduction
N
NXG, however, is more than a disease of the integumentary system. It is, in fact, a systemic variant of adult onset xanthogranulomatous disease and, thus, belongs to a spectrum of xanthogranulomatous diseases, including Erdheim-Chester and adult-onset xanthogranuloma with asthma. NXG is unique within this group because of its systemic and neoplastic effects, which require treatment considerations that can manage the numerous organs involved. Over 80% of patients can develop a paraproteinemia and in such patients the rate of progression to multiple myeloma is 1% annually. 1 Common malignancies associated with NXG include leukemia and lymphoma, as well as blood dyscrasias. 7
While case reports describing periorbital involvement of NXG are frequent, there are only a few case reports describing ocular involvement, such as scleritis and uveitis.8,9 Occasionally, dermatologic involvement may be the initial presentation, sometimes for years, before the development of ocular inflammation. 8 Others, however, have described scleritis before the development of periocular skin lesions.9,10 The present case describes an atypical presentation: that of a granulomatous panuveitis as the presenting feature of NXG with subsequent development of periorbital and upper extremity skin lesions and ultimate eventuation to significant systemic manifestations.
Case
A 36-year-old Armenian female presented to our clinic with a 6-year history of uveitis. Her past medical history was significant for a positive tuberculin skin test before her uveitis development and without any systemic features of infection. She received treatment for latent tuberculosis with a course of isoniazid therapy. At her presentation in our clinic, visual acuity was 20/20 OU. She exhibited a bilateral diffuse anterior and posterior scleritis. There were no features of intraocular involvement. Diagnostic testing (including rheumatoid factor, anti-CCP, ACE, lysozyme, RPR, FTA-ABS, ANCA, and chest X-ray) was unremarkable. She was started on oral prednisone 1 mg/kg/day, which was tapered over several months, but her inflammation recurred at doses lower than 10 mg PO daily. She was subsequently commenced on azathioprine, but failed to respond to this and was continued on oral prednisone. When she became pregnant with her second child her scleritis went into a drug-free remission. She was lost to follow-up for a year and then presented again to our clinic with active bilateral anterior and posterior scleritis with decreased vision (20/50 OD and 20/40 OS). She also exhibited bilateral iris nodules and granulomatous keratic precipitates (Fig. 1) in addition to bilateral 1+ anterior chamber cell and 1+ vitreous haze. Funduscopic examination revealed bilateral choroidal effusions and B scan ultrasonography revealed scleral thickening and choroidal effusions OU (Figs. 2 and 3). She was commenced on oral prednisone 60 mg PO daily, but again was unable to achieve doses lower than 10 mg PO daily without recrudescence of her inflammation. She failed methotrexate 20 mg PO weekly, as well as mycophenolate mofetil 1.5 g PO BID, due to efficacy. Adalimumab 40 mg subcutaneous q 2 weeks was commenced, but due to persistent disease activity, she was advanced to weekly dosing. Despite this advancement, her uveitis and scleritis failed to respond and she required high doses of prednisone to manage her inflammation.

Granulomatous keratic precipitates with atypical flat appearance following the use of topical corticosteroids.
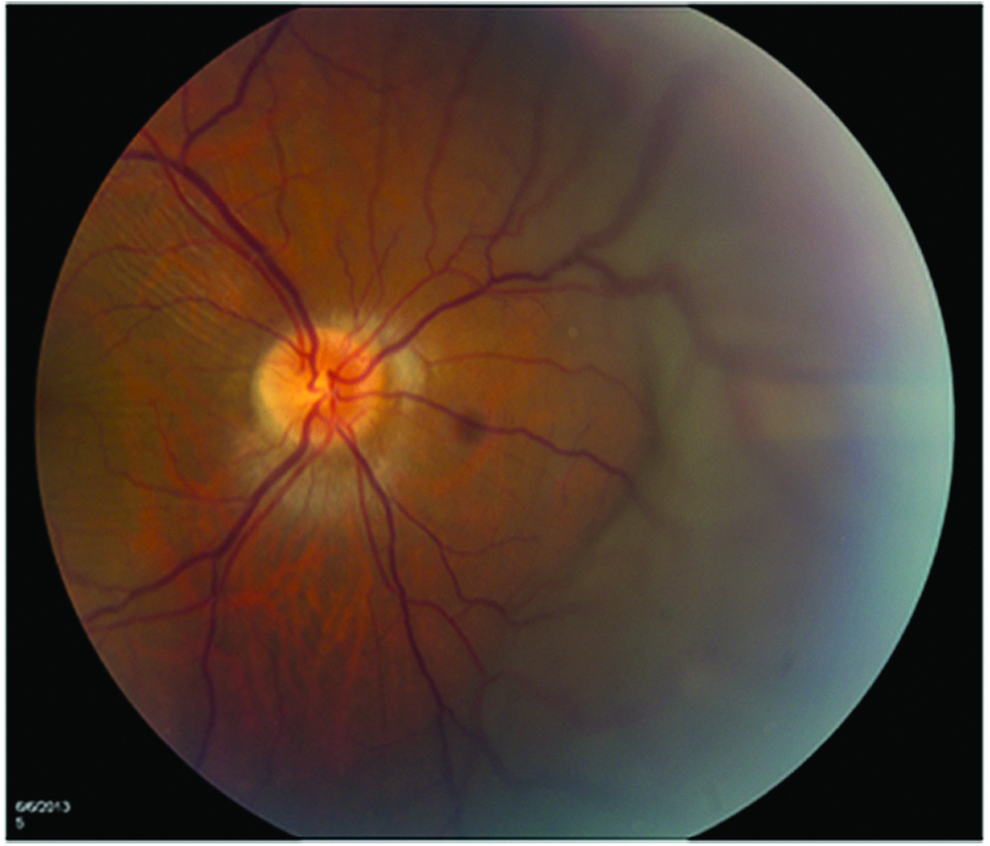
Fundus photo of the right eye with diffuse choroidal effusion nasal to the disc and choroidal folds involving the macula.
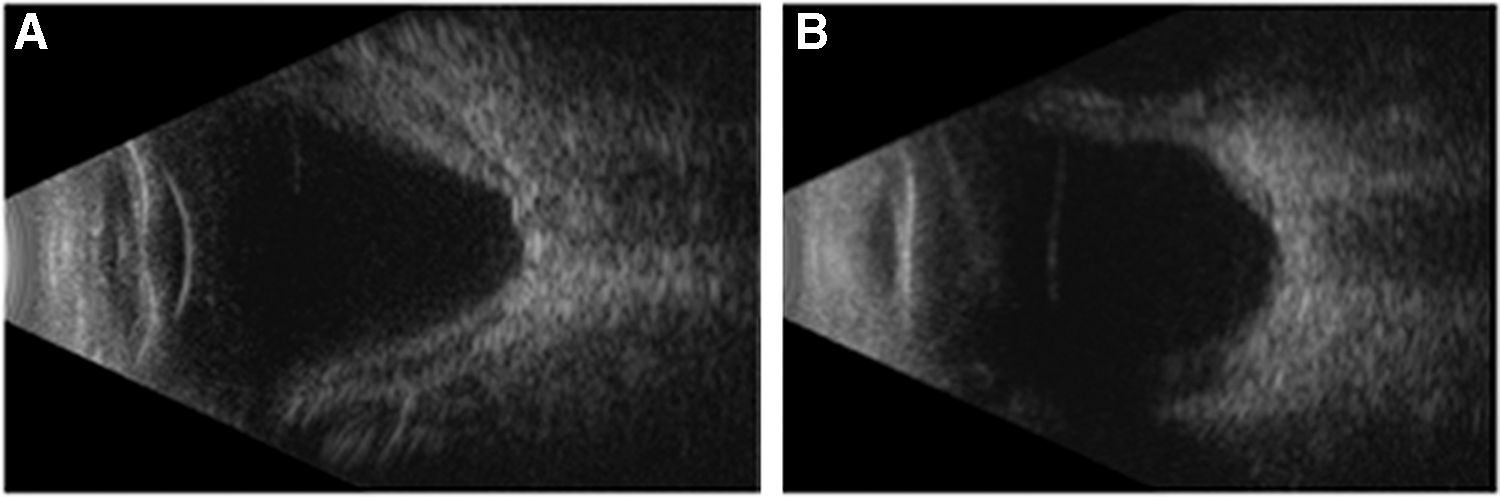
B scan OD
In the interim, she developed systemic symptoms. Paresthesias and a petechial rash involving her lower extremities developed. A biopsy of the lower extremity rash showed microthrombi in the blood vessels, concerning for a Type I cryoglobulinemia. In addition, she became neutropenic (absolute neutrophil was as low as 200 cell/μL) and thrombocytopenic (ranging from 150,000 to 80,000/μL). Peripheral blood smear was normal, and a bone marrow biopsy revealed moderate hypercellularity, but normal hematopoiesis, and no features of a lymphoproliferative disorder. However, there were foci of hemophagocytosis with erythrocytes and neutrophils identified in rare marrow histiocytes. Abdominal ultrasonography showed hepatosplenomegaly. Her ocular and systemic features were at least partially compatible with tuberculosis or sarcoidosis, but rheumatologic, infectious disease, and hematologic consultations did not find any evidence of tuberculosis and sarcoidosis, as a computed tomography of the chest and thorax was negative.
The patient went on to develop periorbital lesions characterized by induration and a yellowish hue (Fig. 4). Initially, they were small, but within 4 weeks became larger with some becoming confluent. In addition, she developed subconjunctival xanthochromic plaque-like lesions, most concentrated at the border with the cornea's superior limbus (Fig. 5). Excisional biopsy of some of these subconjunctival lesions from the left eye was submitted for flow cytometry with no evidence of a lymphoproliferative disorder. One of the lesions involving her right superior eyelid spontaneously ulcerated (Fig. 6). More periorbital lesions developed prompting a magnetic resonance imaging of the orbits, which revealed bilateral lacrimal gland enlargement and bilateral scleral thickening (Fig. 7). Biopsies of the lesions involving the right upper eyelid, left conjunctiva, right orbital fat, and right lacrimal gland revealed non-necrotizing granulomatous infiltrates. There was no evidence of a lymphoproliferative disorder by flow cytometry, and no organisms were seen on gram and acid fast stains.

Xanthelasmic plaque-like lesions in the subconjunctival space, especially dense at the superior corneal limbus.
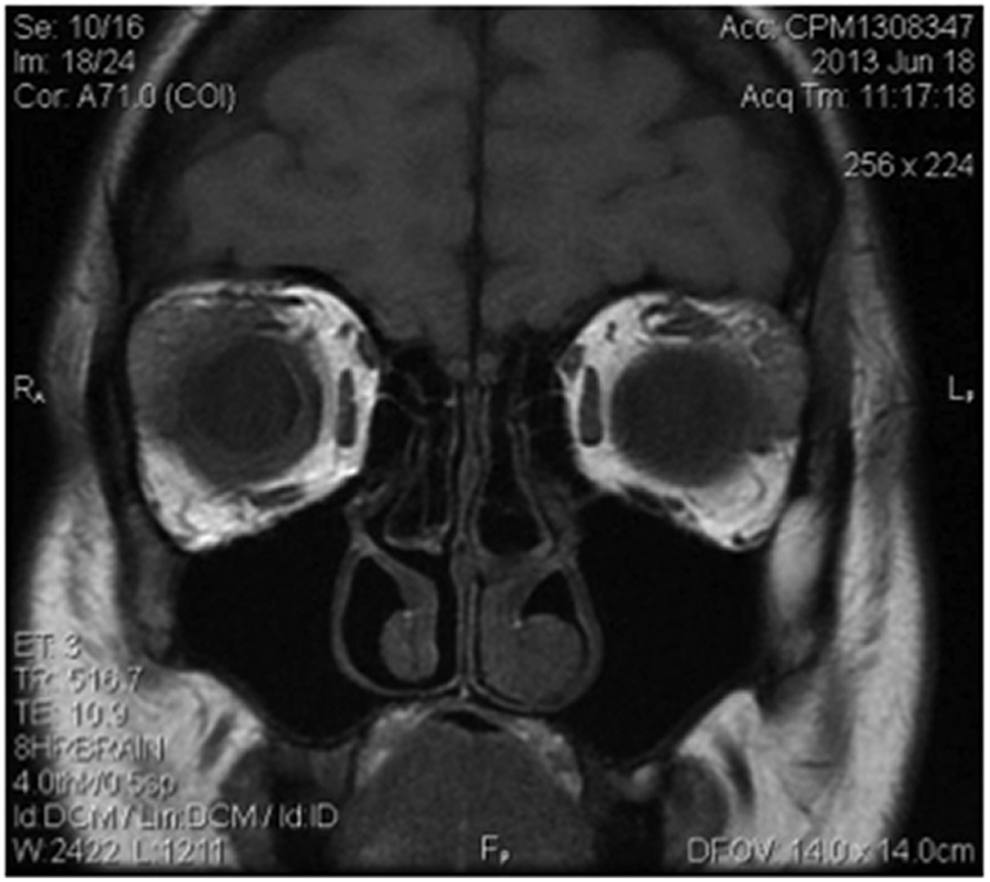
MRI orbits demonstrating bilateral lacrimal gland enlargement and scleral thickening. MRI, magnetic resonance imaging.

Periorbital xanthelasmic-like lesions with healed ulcerative lesion over the right upper eyelid.

Periocular skin lesions.
As her biopsy and clinical ocular findings were most consistent with sarcoidosis, and, given her failure with oral antimetabolites and a tumor necrosis factor (TNF) inhibitor, rituximab was commenced due to active scleritis. She received 2 cycles of intravenous rituximab 1,000 mg IV over 2 weeks with 60 mg of oral prednisone daily. She was then continued on oral prednisone, but as she required high doses of this, methotrexate 20 mg subcutaneously every 7 days was started. Shortly after, the patient developed arthralgias and joint effusions, which were considered to be related to methotrexate, and this was discontinued. She was restarted on oral prednisone 60 mg PO daily and received posterior sub-Tenon's triamcinolone injections for her choroidal effusions. The patient's bilateral scleritis and panuveitis responded well to the high doses of oral prednisone and sub-Tenon's triamcinolone injections.
The patient then became febrile (up to 103°F), developed diffuse arthralgias, and was hospitalized in acute renal failure (creatinine increased from 0.7 to 2.7 mg/dL). Repeat diagnostic testing, including ANA, c-ANCA, p-ANCA, ACE, lysozyme, and lupus anticoagulant, was unremarkable. A renal biopsy revealed a membranoproliferative glomerulonephritis with thrombotic microangiopathy (due to platelet–fibrin thrombi in arterioles and glomerular capillaries). There was interstitial nephritis featuring chronic inflammation characterized by lymphocytes, macrophages, and eosinophils with interstitial edema as noted on trichrome stain. There was no staining for IgM, IgA, IgG, C3, fibrinogen, or kappa or lambda light chains. No immune deposits were noted on electron microscopy, but widespread foot process effacement was seen. Perhaps most interesting were droplets that were seen on trichrome and PAS in some of the glomerular capillary loops, suggesting a cryoglobulinemia. Indeed, serum protein electrophoresis showed an IgG monoclonal gammopathy. In light of these findings, a full-body computed tomography-positron emission tomography scan was performed and was negative for a systemic neoplastic process.
Ultimately, the patient developed a new indurated lesion on her left shoulder, which was biopsied. Histopathology of this biopsied lesion revealed bizarre shaped multinucleated giant cells (Touton type) with palisading histiocytes around disorganized collagen (zones of necrobiosis), as well as characteristic cholesterol clefts (Fig. 8). These features were classic for NXG.

Histopathology of left shoulder lesion skin biopsy. This shows multinucleated giant cells (detail, inset
The patient was ultimately placed on a regimen of monthly intravenous immunoglobulin (IVIG) 0.4 mg/kg in addition to thalidomide 50 mg PO daily, cyclophosphamide 100 mg PO daily, and prednisone 12.5 mg PO daily. She has exhibited no further progression of her periocular lesions and is systemically stable. In addition, her uveitis has been inactive. Her remaining issue is that of exposure keratitis in the right eye due to poor lid closure; this is being managed with aggressive lubrication.
Pathogenesis
The pathogenesis of NXG is unknown. Some have considered an infectious etiology, such as Borrelia burgdorferi, 11 to be at play in xanthogranulomatous processes, 12 but infectious DNA has rarely been found. The xanthomatous nature of the subcutaneous lesions has prompted some to consider a hyperlipemic disorder at play, but the majority of patients with NXG are normolipemic or hypolipemic. 13 Interestingly, 1 patient with NXG exhibited peripheral blood monocytes with higher levels of cholesteryl ester compared to a control, suggesting the accumulation of lipid by peripheral blood monocytes. This seemed to be related to enhanced phagocytic activity of the patient's monocytes. 14 It has also been postulated that an immune response develops due to the paraproteinemia with subsequent deposition of monoclonal protein in tissues. Given that monoclonal gammopathy (IgG) is found in the majority of NXG patients, it is thought that this paraprotein plays a role in the development of the unique granulomas by non-Langerhans histiocytes (thus, nonantigen presenting cells) found in the skin. 15
Diagnosis
Biopsy of involved tissues is the gold standard. Hematoxylin and eosin staining is ideal for identifying granulomas with zones of necrobiosis and cholesterol clefts. These areas will demonstrate infiltration with Touton cells, lymphocytes, and epithelioid histiocytes. Plasma cells may form aggregates as well. Staining for lipids should be supplanted by immunohistochemical stains. Recently, Saggini and colleagues described the use of immunohistochemical staining for adipophilin in the identification of 2 cases of NXG. 16 Similar patterns of staining can be seen in a benign condition, necrobiosis lipoidica, that NXG mimics. 17 However, NXG has much more diffuse adipophilin staining compared to necrobiosis lipoidica. 16
Differential diagnosis
Initially, NXG may have protean systemic or ocular inflammatory presentations, which can delay the diagnosis, and, thus, the institution of appropriate therapy. Xanthomatous lesions such as xanthelasma frequently involve the periocular skin and eyelids and can mimic the cutaneous lesions seen in NXG as they too may have a yellowish hue. The lesions in NXG, however, tend to be much deeper and firmer than xanthelasmas and NXG lesions can spontaneously ulcerate, which does not occur in xanthelasmas. Histopathologically, the xanthomatous lesions are characterized by foamy lipid laden macrophages within the dermis. 18 Necrobiosis lipoidica diabeticorum can also masquerade as the cutaneous lesions in NXG, although it tends to favor the legs; biopsy in necrobiosis lipoidica diabeticorum shows an absence of cholesterol clefts 8 and confinement to the dermis, whereas NXG frequently involves the subcutaneous region. 1 Sarcoidosis can also involve the skin (including periocular regions), but this only occurs in up to 35% of sarcoid cases. 19 Cutaneous sarcoidosis in the facial region may appear as red-brown to violaceous papules and plaques (in contrast to the yellow hue of NXG), often studding the nasal alar rims (lupus pernio).19–21 Subcutaneous nodular lesions can also occur in sarcoidosis with a classic location being the extensor forearms. 22 As sarcoidosis most frequently affects the lungs 23 identifying pulmonary involvement on radiographic imaging can delineate tissue amenable for biopsy. Pulmonary involvement in NXG, on the other hand, occurs much less frequently than in sarcoidosis and lacks the typical radiographic features used to stage pulmonary sarcoidosis. 24 In addition, while polyclonal hypergammaglobulinemia can be seen in sarcoidosis,25–27 monoclonal paraproteinemias (as in NXG) are not frequently encountered. Thus, in a patient with granulomatous uveitis and scleritis, the development of a paraproteinemia may suggest a cause other than sarcoidosis. Another condition that should be considered in the differential diagnosis is hemophagocytic lymphohistiocytosis (HLH). 28 In HLH macrophages secrete high amounts of pro-inflammatory cytokines leading to systemic inflammation and organ damage. The variable manifestations as a consequence of systemic inflammation in HLH can mimic the systemic inflammation seen in sarcoidosis and NXG. In fact, HLH can occasionally occur in association with sarcoidosis. 29 Finally, tuberculosis is known as the great masquerader as it can cause infection and granulomatous inflammation anywhere in the body. Tuberculosis can cause granulomatous ocular inflammation similar to sarcoidosis. In addition, tuberculosis may rarely present in the skin as red-brown plaques (eg, lupus vulgaris), but such lesions lack the yellow xanthomatous appearance of NXG.30–32 Since tuberculosis has a predilection for pulmonary involvement, identifying the radiographic features of tuberculosis in addition to establishing a positive tuberculin skin test or interferon-gamma release assay will aid in securing a diagnosis for this infectious entity. Another masquerader, syphilis, can also cause ocular inflammation, as well as granulomatous inflammation. Syphilitic gummas may involve the facial skin and can even ulcerate.33,34 However, treponemal testing will help identify cases of syphilis.
Treatment of NXG
There are no well-defined treatment guidelines for NXG especially with concurrent ocular involvement. Often, a multimodal and multidisciplinary approach is necessary when multiple organs are involved and in the context of lymphoproliferative disorders and blood dyscrasias.
Topical
In the case of uveitis and scleritis, the location of inflammation will dictate therapy. For anterior uveitis, topical corticosteroids (eg, prednisolone acetate 1%) can be effective. Indeed, in our patient, topical prednisolone acetate 1% managed the anterior chamber features of uveitis well. Mild anterior scleritis may be managed with topical corticosteroid drops as well, but may require systemic therapy for cases that are more aggressive.
Intralesional/periocular
Intralesional injection of triamcinolone acetonide has been considered particularly for the periocular lesions in NXG. 35 Indeed, in our case, we gave an intralesional injection of 0.2 mL of 40 mg/mL of triamcinolone acetonide into a periocular skin nodule, which resulted in an undesirable result of darkening of the overlying skin. Others have noted a lack of efficacy as well. 36 However, our patient responded well to posterior sub-Tenon's triamcinolone acetonide (1 mL of 40 mg/mL) injections (both the Nozik technique, 37 as well as inferior orbital floor approach) particularly when her scleritis resulted in serous choroidal detachments. Subconjunctival triamcinolone acetonide may be used for non-necrotizing scleritis in general, although in NXG necrotizing scleritis can characterize the scleral inflammation 8 and in such cases, subconjunctival corticosteroids should not be used.
Systemic Treatment Options
Corticosteroids
Most cases of the dermatologic involvement in NXG have been treated systemically and we found this to be the most useful strategy for the ocular inflammation as well. The mainstay of systemic therapy in NXG is systemic corticosteroids, which may be used alone or in combination with other systemic therapies, topical therapy, or radiation. Systemic corticosteroids are frequently effective in NXG manifesting with periocular involvement,36,38 but the utility when NXG is associated with scleritis and uveitis is not as well known. Despite this, approaching ocular inflammation in NXG as is done for any other noninfectious cause of uveitis or scleritis is reasonable. We suggest using doses of prednisone 1 mg/kg/day, although doses above 60 mg PO daily are not recommended. 39 For particularly robust inflammation compromising visual acuity, intravenous methylprednisolone 1,000 mg daily may be considered for 3–5 days before converting to oral prednisone. The goal should be to decrease the systemic corticosteroids in a stepwise manner ensuring that the inflammation is controlled as dose reductions are made. It would be reasonable to consider long-term therapy with prednisone (or an equivalent corticosteroid) dose of 7.5 mg PO daily or less. However, if the inflammation should require doses of corticosteroids more than what is considered physiologic, steroid-sparing therapy should be sought out given the side effects of moderate-to-high doses of systemic corticosteroids. Unfortunately, it is frequently difficult to taper to low doses of corticosteroids.
Antimetabolites
Antimetabolites are a reasonable option for first-line corticosteroid sparing therapy of NXG-related uveitis and scleritis. Antimetabolites have been shown to be effective in the management of ocular inflammatory diseases,40–42 although randomized prospective trials assessing the efficacy of antimetabolites for uveitis are very recent. 43 Thus, in the even more infrequent case of NXG, efficacy of the most commonly used antimetabolites used for ocular inflammation (methotrexate, azathioprine, and mycophenolate mofetil) is entirely unknown. Methotrexate and azathioprine have been used for NXG, but may not always provide a satisfactory response with respect to management of subcutaneous nodules, uveitis, or systemic manifestations. While others have noted a good response to methotrexate in the setting of adult-onset orbital xanthogranuloma, 44 for example, it is unclear whether antimetabolite therapy alone is sufficient for controlling the systemic features in NXG let alone its associated ocular inflammatory disease.
Thalidomide
Thalidomide 45 and lenalidomide 46 are frequently used for dermatologic involvement in NXG. Thalidomide affects tumor necrosis factor alpha (TNF-alpha). 47 Lenalidomide, which is a derivative of thalidomide, has been used more commonly as a chemotherapeutic involved in neoplastic cell apoptosis. Lenalidomide seems to exert at least part of its action by affecting regulatory T cells. 48 Some of the rare cases of posterior scleritis have used thalidomide with success. 9 Importantly, thalidomide and lenalidomide show promise in treating multiple myeloma when it is resistant to melphalan and may have benefit in NXG by reducing pathogenic paraproteins. 49 There is little experience using thalidomide or lenalidomide for uveitis specifically. A single case report describing uveitis related to lepromatous leprosy documented response to thalidomide, 50 as well as a child with chronic bilateral uveitis failing periocular steroid injections, as well as systemic cyclosporine. 51 Animal models of uveitis have demonstrated effectiveness of thalidomide in controlling ocular inflammation.52–54 Thalidomide and lenalidomide are used with success in Behçet's disease, particularly when oral and genital ulcerations are severe.55–57
Alkylating agents
This group of medications includes chlorambucil,58,59 cyclophosphamide, 60 and melphalan36,38,61 and can be considered in the event that therapy with antimetabolites or thalidomide/lenalidomide is not effective. By alkylating and cross-linking DNA of rapidly dividing inflammatory cells, the theoretic response in NXG should be a decrease and improvement in the subcutaneous nodules and ocular inflammation. However, because these medications can have profound side effects, this class of agents is typically reserved for severe uveitis or scleritis where blindness is probable. Indeed, our patient's robust ocular flares were an indication to utilize cyclophosphamide.
Dapsone
Dapsone, an inhibitor of dihydrofolic acid synthesis, has been used in NXG with varying success.62,63 While dapsone has been used with reasonable success in ocular inflammatory disorders (such as cicatricial pemphigoid 64 ), its use in the setting of NXG-related uveitis or scleritis is not known.
Biologics
Interferon alpha-2a 65 and interferon alpha-2b 66 have each been used in 2 patients, respectively, although only the patient receiving interferon alpha-2b had periocular lesions (none with uveitis or scleritis). The frequency with which patients may achieve a durable remission, especially compared to alkylating agents and antimetabolites, is unknown. Moreover, the response of ocular disease, particularly scleritis or uveitis, in the setting of NXG is not known. Another biologic medication, infliximab (a TNF-alpha inhibitor), has been used in a case of NXG with involvement of the periocular skin. 67 This medication had to be discontinued, however, due to recurrent infections. While anti-TNF agents are frequently used in uveitis, they also demonstrate efficacy in multiple myeloma.68–70 Inhibiting TNF may be a reasonable therapeutic approach in NXG where multiple myeloma is a complication. Rituximab, an anti-CD20 biologic that depletes B-cells, is an important new therapy for refractory scleritis,71,72 although its use in scleritis in NXG has not been reported before this report. In the present case, however, anti-TNF-alpha therapy and rituximab were not effective in managing the systemic and ocular inflammation.
IVIG has been used either alone61,73 or in combination with photophoresis 67 or radiation 74 with relatively good success. However, only 2 of the reported patients had periocular dermatologic involvement61,73 (in addition to systemic features) and none of these cases had frank scleritis or intraocular involvement.
Surgical
Surgical excision may be considered for periocular subcutaneous nodules. However, this should only be considered when the inflammation is controlled lest new lesions develop at a later time. One concern is that, similar to ocular cicatricial pemphigoid, surgical intervention may promote an inflammatory generating environment that can lead to further nodule or scar formation. Indeed, surgical excision alone without systemic therapy has resulted in recurrence of periocular lesions. 75
Radiation
Radiation has been suggested as an option for treating the periocular subcutaneous nodules in NXG 76 with doses of 1,500–2,400 cGy being typically used. 38 Whether radiation following control of inflammation with systemic immunomodulatory or biologic therapy may be more effective in terms of preventing recurrences of periocular disease compared to radiation alone is unknown.
Our recommendations
Given the extreme rarity of NXG, and the even less frequent ocular involvement (in the form of scleritis, uveitis, or both), treatment can be challenging at best and bewildering at worst. It is important to appreciate the utility of systemic corticosteroids. However, it is equally important to recognize that a corticosteroid-sparing agent will be necessary due to the chronicity of the disorder and the hematologic and lymphoproliferative features that characterize NXG and differentiate it from the other xanthogranulomatous diseases (Table 1). Antimetabolites are a reasonable option to begin with, but whether another antimetabolite should be attempted when a particular one fails is unknown. In the period of time that it takes for a particular antimetabolite to reach therapeutic efficacy (typically at least 3 months) there may have been further progression of the disease and the physician and patient will have to decide whether it is likely that another antimetabolite will also be ineffective. First- or second-line therapy with thalidomide (or its derivative, lenalidomide) should be considered particularly when multiple myeloma complicates NXG. In addition, alkylating agents may be used as third-line therapy either alone or in combination with thalidomide or lenalidomide as was done in the present case to achieve control of systemic and ocular inflammation. Whether biologics (including TNF inhibitors or anti-CD20 agents) have utility in the ocular inflammation that can complicate NXG remains to be determined. The uveitis and scleritis in the present case did not respond to biologics suggesting inflammatory mediators distinct from other types of noninfectious uveitis and scleritis. Tailoring a particular therapy for a patient will require close collaboration among the specialties of uveitis, oculoplastics, rheumatology, hematology-oncology, and dermatology. Physicians and patients should be prepared that first-line medications may not control ocular inflammation and advancement of therapy may be required. For example, in a series of 35 patients with NXG and concurrent paraproteinemia (66% having periocular dermatologic involvement), the median number of different types of therapy tried before achieving control of inflammation was 3 7 indicating the lack of a well-defined systemic therapeutic approach. Thalidomide and lenalidomide were most frequently used followed by alkylating agents.
Complications in NXG
A significant complication of NXG is delayed diagnosis. Given the systemic manifestations of the disease, particularly when skin, subconjunctival, or orbital lesions are not especially prominent (or amenable to biopsy) initially, a specific diagnosis can be elusive. Bone marrow biopsy is often used when there are blood dyscrasias. The initial skin biopsies in our patient did not reveal zones of necrobiosis, which delayed the diagnosis of NXG. In addition, our patient's bone marrow biopsy revealed a hemophagocytosis. In the context of this patient's splenomegaly, thrombocytopenia, and neutropenia, the differential diagnosis could have been compatible with HLH 77 if it were not for her skin involvement that ultimately disclosed NXG. This is an interesting feature that we have not encountered in the bone marrow biopsy features of other patients with NXG described in the literature.
Lagophthalmos as a result of periocular involvement can result in exposure keratopathy with subsequent perforation, endophthalmitis, and loss of the eye. 78 In our patient, lagophthalmos and poor blink reflex in the right eye have led to exposure keratitis requiring frequent lubrication. Exposure keratopathy puts this patient at risk for infectious keratitis. The disfigurement of the face in NXG patients can be a significant concern and oculoplastic surgery may be of benefit in achieving a satisfactory outcome for a patient. 79
Patients with NXG must be monitored for the development of or worsening of existing blood cell and plasma cell dyscrasias and lymphoproliferative processes. In addition, since NXG is thought to be a chronic condition, patients may require long-term systemic immunosuppression. Whether or not systemic immunosuppression decreases the frequency of developing plasma cell dyscrasias is not known.
The present case illustrates ocular inflammation (scleritis and uveitis) presenting years before dermatologic and systemic involvement in NXG. When new systemic features declare themselves, it is important to reassess whether the ocular inflammation is truly an independent process or part of a larger inflammatory process. Future studies are necessary to identify ideal therapeutic approaches for both the ocular and systemic inflammation in NXG.
Footnotes
Author Disclosure Statement
No competing financial interests exist.