
Abstract
Purpose:
This study investigated the protective effect of Kaempferol against hydrogen peroxides (H2O2)-induced retinal pigment epithelium (RPE) cell oxidative stress, inflammation, and apoptosis and investigated if this protection involves modulation of poly(ADP-ribose) polymerase-1 (PARP1)/silent information regulator 1 (SIRT1) signaling pathway.
Methods:
ARPE-19 cells were pretreated with increasing doses of Kaempferol (10, 25, 50, 100 μM) for 24 h in Dulbecco's modified Eagle's medium/F-12 medium with or without postincubation with H2O2. Control cells remained untreated.
Results:
Kaempferol, in a dose-dependent manner, significantly increased cell survival and reduced levels of reactive oxygen species, malondialdehyde, single-stranded DNA (ssDNA), and lactate dehydrogenase but increased levels of glutathione (GSH) and manganese-superoxide dismutase (MnSOD) in H2O2-treated ARPE-19 cells. It also increased GSH and MnSOD in a dose-dependent manner in control + Kaempferol treated cells. At a dose of 50 μM, the most effective dose, Kaempferol also inhibited protein levels of tumor necrosis factor alpha and interleukin-6, nuclear activity and protein levels of total, acetylated, and cleaved PARP1, and increased nuclear levels and activity of SIRT1 in H2O2-treated cells. In parallel, it increased total nuclear levels of Nrf2 but reduced the acetylation of p53, Nrf2, nuclear factor-κB (NF-κB) p65, and forkhead transcriptional factor 1 (FOXO1). Of interest, the stimulatory role of Kaempferol in the nuclear accumulation and activation of SIRT1 and the nuclear levels of Nrf2, as well as in reducing the acetylation of Nrf2, NF-κB p65, and FOXO1, was shown in nuclei of control + Kaempferol-treated cells.
Conclusion:
Kaempferol protective effect against H2O2-induced ARPE-19 damage involves antioxidant and anti-inflammatory effects mediated, at least, by stimulating the nuclear accumulation, activation, and deacetylase ability of SIRT1 and concurrent inhibition of PARP1.
Introduction
Age-related macular degeneration (AMD) remains one of the most common retinal diseases in elderly people, and is associated with vision problems and blindness. 1 The prevalence of AMD among middle age and old people is significantly increasing, and recent data have shown that ∼30% of the AMD patients are visually impaired.1–3 The retinal pigment epithelium (RPE) cells are a single layer of epithelial cells that are located in the outer layer of the retina, just between the choriocapillaris and photoreceptors, and form a part of the blood–retina barrier. 4 These cells have a high metabolic and oxygen consumption rate, and play an essential role in the regeneration, repair, and viability of the photoreceptor, as well as in suppressing the neovascularization and retinal edema.5–7 For this reason, the RPE cells are usually exposed to high levels of reactive oxygen species (ROS), which create an oxidative stress response that leads to activation of inflammation and apoptosis in these cells.7–9 Currently, it is well accepted that overproduction of ROS (e.g., superoxide, hydroxyl radicals, singlet oxygen, and hydrogen peroxide (H2O2)) is the leading cause of RPE cell damage in AMD patients and animal models, specifically in those of the nonexudative dry type.7–10
On the contrary, the contribution of the 2 related enzymes, poly(ADP-ribose) polymerase-1 (PARP1) and the silent information regulator 1 (SIRT1), to the regulation of retinal cells morphology, function, and survival continued to fascinate scientist over the last decades.11,12 SIRT1 is an NAD+-dependent protein deacetylase that promotes cell survival through stimulating the synthesis of the antioxidant enzyme and inhibition of cell inflammation and apoptosis through deacetylation of several transcription factors, including p53, nuclear factor-κB (NF-κB), peroxisome proliferators-activated receptor-γ (PPAR-γ) and its coactivator-1α (PGC-1α), and forkhead transcriptional factors (FOXO). Mechanisms behind this are well explained in some previous studies.13–15 Furthermore, recent evidence has shown that antioxidant potential of SIRT1 is mediated by the upregulation and nuclear deacetylation of the nuclear factor erythroid 2-related factor transcription factor (Nrf2), which normally stimulates the transcription of a variety of antioxidant genes through bindings to the antioxidant response element.16–18 Also, Nrf2 has been described as an anti-inflammatory agent that can inhibit NF-κB, and the subsequent synthesis and release of many inflammatory cytokines, thus suggesting that SIRT1 anti-inflammatory effect is also mediated by deacetylation/activation of Nrf2.19,20 However, PARP1 is the most common DNA repair enzyme in most cells, which is usually found in the nuclei and mitochondria fractions and is activated in response to DNA damage to promote the NAD+-dependent protein poly(ADP-ribosyl)ation (PARylation) and DNA repair. 21 However, while a transient activation of PARP1 was shown to promote cell survival, sustained activation of PARP1 can induce cell apoptosis by several mechanisms including energy failure (decreasing ATP stores) and antagonizing the effect of SIRT1 through depletion of inhibition of cellular levels of NAD+.21–24
Nevertheless, localization studies have shown that SIRT1 is normally expressed in rodents' cornea, lens, ciliary body, iris, and throughout the retina cells, including inner, outer, and ganglion layers in abundant quantities. 25 Expression and activity of SIRT1 were significantly reduced in the retina and capillary cells in different ocular degenerative disorders such as diabetic retinopathy and glaucomatous neurodegeneration and other conditions including cataract, uveitis, and optic neuritis. 11 However, in the majority of these studies, the upregulation/activation of SIRT1 afforded protection. 11 Also, low levels of SIRT1 were reported in retinal cells in AMD patients and rats after light-induced retinal damage, whereas activation of SIRT1 by resveratrol protected the retina structure and function.26,27 Besides, the activation of SIRT1 by pharmacological agents protected against amyloid beta-induced changes in cell morphology and damage of human RPE cells by decreasing the acetylation and inhibition of NF-κB. 28
Opposing the low level of SIRT1, elevated expression, and activation of PARP1 were shown in the retina cells in a variety of retinal degeneration conditions such as retinitis pigmentosa and diabetic retinopathy, as well as after ischemia/reperfusion, hypoxia, and oxidative stress-induced RPE death.29–34 However, inhibition of PARP1 protected the retina cells from oxidative damage by stimulation of different mechanisms, including activation of protein kinase B (Akt) and the nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor, as well as through inhibition of JNK, P38 mitogen-activated protein kinases (P38 MAPKs), and NF-κB.31–34
Nonetheless, the plant's flavonoids glycoside, Kaempferol (3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-1-benzopyran-4-one), is currently under extensive research as a potent protective agent against oxidative and inflammatory damage with many thanks to its potent antioxidant, anti-inflammatory, and antiapoptotic properties. 35 As a protective flavonoid in the retina, Kaempferol inhibited angiogenesis in human retinal endothelial cells by downregulation of Src-Akt1-Erk1/2 signaling pathway, vascular endothelial growth factor (VEGF), and placenta growth factor. 36 Also, Kaempferol enhanced cell survival and protected the human RPE cells from H2O2-induced oxidative damage and apoptosis by suppressing ROS generation, downregulation of VEGF, and upregulation of superoxide dismutase (SOD). 37 In extra-retinal tissues, Kaempferol protected against H2O2-induced apoptosis in liver and lung cells.38–40 Of interest, Kaempferol also inhibited PARP1 and reduced cytokines production in lipopolysaccharide-stimulated pulmonary epithelial cells. 40 These data support the further hypothesis that Kaempferol may protect the RPE cells from H2O2-induced oxidative damage by inhibition of PARP1, possibly through increasing the nuclear accumulation of SIRT1.
Therefore, in this in vitro study, we aimed to investigate the protective effect of Kaempferol against H2O2-induced oxidative stress, inflammation, and apoptosis in RPE cells, and to investigate if the mechanism of protection involves modulation of the activity and levels of PARP1/SIRT1 axis and SIRT1 downstream targets.
Materials
Cell line and culture
The human RPE cell line (ARPE-19) was purchased from the American Type Culture Collection. Cells were cultured at 37°C in an atmosphere of 5% CO2 in Dulbecco's modified Eagle's medium/F-12 medium (Corning, Tewksbury) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, CA).
Cell treatments
The cells were passaged every 3 days by 0.25% Trypsin/EDTA and used within the first 8 passages at a confluence of 85%. For the experimental procedure, ARPE-19 cells were seeded in 96-well plates at a density of 5 × 104 cells/well and were pretreated with increasing concentration of Kaempferol (10, 25, 50 μM) (Cat. No. 60010; Sigma Aldrich, St. Louis, MO) for 24 h, and then incubated or not incubated with H2O2 (300 μM) for the next 24 h. After this part, cell viability, levels of lactate dehydrogenase (LDH) in the media, and levels of ROS and single-stranded DNA (ssDNA) in all cell treatments were determined as explained below. Based on these results, the most effective nontoxic dose of Kaempferol was used for the rest of the experimental procedures. Kaempferol was always prepared in dimethyl sulfoxide (DMSO) and then diluted with phosphate-buffered saline (PBS; final concentration of DMSO was <0.05%). Control cells were treated only with the DMSO. In our preliminary data, DMSO (0.050%) was shown to be nontoxic, having no effect on cell count, ROS levels, and LDH release in control cells. The dose of H2O2 was adopted from the study of Chang et al. 41 All experiments in this study were done in triplicate and ∼3 trails/groups.
Determination of cell viability
The cell counting kit-8 (CCK-8, Cat. No. CK04-13; Dojindo, Kumamoto, Japan) was used to determine cell viability following the manufacturer's instruction. In brief, 100 μL of the medium of all wells were removed and replaced with a fresh medium containing 10% of CCK8 solution. After 3 h incubation at 37°C, the absorbance of all samples was determined using a microplate reader at 450 nm using a Spectramax M2 plate reader (molecular devices, CA). Calculation of cell viability as a percentage was determined by using the following equation [(Abs of the sample − Abs of the blank) / (Abs of the control − Abs of the blank)].
Determination of LDH levels
Media levels of LDH were measured using a commercially available colorimetric kit (Cat. No. ab102526; Abcam, Cambridge, United Kingdom) per the manufacturer's instruction. The test relies on the reduction of NAD+ to NADPH, which then interacts with a specific probe to produce a color that can be read at 450 nm. In the test, a 20 μL of each sample (adjusted to 50 μL with the LDH assay buffered) were mixed with 50 μL of the reaction mixture (48 μL LDH assay buffer and 2 μL LDH substrate buffer) and read immediately at 450 nm on Spectramax M2 plate reader (molecular devices). LDH concentrations were measured using a standard curve.
Determination of apoptosis by ssDNA assay
The levels of ssDNA, as markers of cell apoptosis and DNA damage, were determined using a commercially available kit (Cat. No. APT225; Millipore) following the manufacturer's instruction. In brief, cells of each sample were centrifuged at 112,000 g for 5 min, and the pellet was collected. The cells were fixed with 80% methanol, which was then removed, and the plate was dried in a water bath at 37°C for 30 min. Then, 50 μL formamide solution were added to each well and incubated at room temperature for 10 min, and then at 75°C for another 10 min. After that, the tube was allowed to cool, and the formamide solution was discarded. Then, 200 μL of 1% bovine serum albumin (BSA) were added to each well to block any nonspecific binding. After 5 min, the excess BSA was removed, and all wells were incubated with 50 μL mouse monoclonal anti-ssDNA antibody (1:100) and 200 μL horseradish peroxidase (HRP)-secondary antimouse secondary antibody (1:100), each of 30 min at room temperature. Then, all wells were washed with the washing solution 3 times (3 min/each) and incubated with 100 μL 2,2′-azino-bis-(3-benzthiazoline-6-sulfonic acid) solution for 30 min at room temperature. Finally, a stop solution was added to all wells, and absorbance was read by Spectramax M2 plate reader (molecular devices) at 405 nm.
Preparation of cell homogenates and nuclear fractions
To prepare total cell homogenates for the biochemical analysis, cells were pelleted and then homogenized in 100 μL of ice-cold PBS (pH of 7.4) supplied with 10 μL protease inhibitor cocktail (Cat. No. P8340; Sigma-Aldrich). The supernatants were collected and stored at −80°C until use. For western blotting experiments, total protein was extracted using RIPA buffer lysis kit (Cat. No. 89900; ThermoFisher) according to the manufacturer's instruction. The nuclear and cytoplasmic fractions were prepared using a cytoplasmic/nuclear extraction kit (Cat. No. 78835 and ThermoFisher Scientific). Protein levels in total, nuclear, or cytoplasmic proteins were determined using a Bradford assay (Cat. No. 23300; ThemoFisher Scientific, MA).
Measurements of ROS
The levels of ROS in all cells were measured spectrophotometrically using a green fluorescence assay kit (Cat. No. ROS0300; OZBioscience). The test measures the quantity of ROS produced in the cells using the cell-permeable fluorogenic probe 2′,7′-Dichlorodihydrofluorescein diacetate (DCF-DA). The principle of the test is based on reading the fluorescent signal of DCF, which is produced intracellularly by the oxidation of the diffused DCF-DA by ROS. In brief, the culture medium was removed, and the cells were gently washed by 100 μL PBS (pH 7.4). Then, the cells were stained by adding 100 μL of diluted DCF-DA solution and incubated for 30 min in the dark at 37°C. After this incubation, extra DCF-DA solution was removed, and the cells were washed again with 100 μL PBS. After that, the PBS was removed and replaced with a new one of the same volume, and the intensity was measured immediately using a microplate fluorescence reader (FL600; Bio-Tek Instruments, Inc., Winooski, VT) at excitation/emission of 485/535 nm.
Other biochemical measurements in cell homogenates
Cell homogenate levels of reduced glutathione (GSH), malondialdehyde (MDA), and manganese-superoxide dismutase (MnSOD) were measured using special colorimetric and ELISA kits (Cat. No. Ab205811 and Cat. No. ab118970; Abcam, United Kingdom, and Cat. No. CSB-E17064h; CUSABIO technology, TX, respectively). All measurements were performed per the manufacturer's instructions.
Biochemical measurements in the nuclear fraction
The nuclear SIRT1 deacetylase activity was measured using a fluorometric Cyclex SIRT1/Sir2 Deacetylase Fluorometric Assay Kit (Cat. No. CY-1151V; Nagano, Japan) with 20 μg protein of the nuclear extract. The fluorescent signal was read using a microplate fluorescence reader (FL600; Bio-Tek Instruments, Inc.) at excitation/emission of 340/460 nm. The nuclear PARP-1 activity was measured with the help of a universal colorimetric assay kit (Cat. No. 4677-096-K; Gaithersburg, MD) using 25 μg protein of the nuclear extract. The nuclear activity of NF-kB P65 was measured using TransAM assay kit (Cat. No. 40596; Active Motif, Tokyo, Japan) and recombinant NF-kB p65 as a standard (Cat. No. 31102; Active Motif) using 20 μg protein of the nuclear extract. For the measurements of PARP1 and NF-kB P65 activity, the absorbance was read at 450 nm using a Spectramax M2 plate reader (molecular devices).
Immunoprecipitation of PPAR-1
This procedure was performed to immunoprecipitate PPAR-1, P53, FOXO1, and NF-κB p65 for further determination of their acetylated levels. For this purpose, an immunoprecipitation kit was used (Cat. No. 206996; Abcam). In brief, 100 μg of the nuclear protein of each sample was mixed with 5 × volume of the lysis RIPA buffer supplied with 2 μL protease inhibitor cocktail (provided with the kit). For different samples, 4 μg of PPAR-1, P53, FOXO1, or NF-κB p65 antibodies (Table 1) or normal rabbit IgG antibodies were added to each sample and mixed overnight with rotation at 4°C. After antibody binding, 25–40 μL of preprepared protein A/G sepharose beads were added to each sample and incubated at 4°C for 1 h. Samples were then centrifuged at 2000 g for 2 min to collect the protein A/G sepharose beads, which were then washed 3 times with the washing buffer followed by removing the supernatants. The beads were allowed to dry and then were loaded with 40 μL 2 × Laemmli loading buffer, boiled for 5 min, and then stored at −80°C for future sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) western blotting.
Antibodies Used in This Study
FOXO-1, forkhead transcriptional factor 1; IL-6, interleukin-6; NF-κB, nuclear factor-κB; PARP-1, poly(ADP-ribose) polymerase-1; SIRT1, silent information regulator 1; TNF-α, tumor necrosis factor alpha.
Western blotting analysis
Equal protein samples (40 μg) were separated on different ratios of SDS-PAGE, transferred onto nitrocellulose membranes, blocked, and blotted with the primary antibodies (Table 1) for 2 h at room temperature with rotation. Then, membranes were blotted against appropriate corresponding HRP-conjugated secondary antibodies for another 2 h at room temperature with rotation. All antibodies were prepared in TBST buffer and applied at a dilution between (1:500 and 1:1000), except for β-actin (1:2000). Bands were developed using a Pierce ECL kit (ThermoFisher, Piscataway, NJ), and their intensities were measured using a C-Di Git blot scanner and software (LI-COR, NE). The intravariation between different gels was normalized using an internal control known sample. All membranes were stripped up to 5 times. For the detection of acetylated P53, NF-κB p65, PARP1, and FOXO-1 in the nuclear fractions, nuclear proteins were first immune-precipitated (as mentioned above), followed by blotting with an acetylated lysine antibody (Cat. No. 9441, 1:500; cell signaling).
Statistical analysis
Statistical analysis for all measured parameters was done using GraphPad Prism statistical software package (version 5&8). Normality was tested using the Kolmogorov–Smirnov test. Differences among the experimental groups were assessed using the Kruskal–Wallis nonparametric analysis. Data were presented as mean ± SD. Values will be considered significantly different when P < 0.05.
Results
Kaempferol increases cell survival and, in a dose-dependent manner, inhibits ROS, LDH release, ssDNA, and MDA in H2O2-treated ARPE-19 cells
In this study, we first tested increasing doses of Kaempferol (10–100) on cell survival, the release of LDH, and levels of ROS, ssDNA, and MDA in both control and H2O2-treated ARPE-19 cells. The cells were initially treated with increasing doses (0, 10, 25, 50, and 100 μM) of Kaempferol for 24 h, and then incubated or not incubated with 300 μM H2O2 for the next 24 h. As shown in Fig. 1A, C, and E, and Fig. 2A and C, none of the tested doses of Kaempferol has any significant effect on all these parameters in control ARPE-19 cells as compared with control-untreated cells. However, preincubating the ARPE-19 cells with H2O2 for 24 h significantly reduced cell survival and increased intracellular levels of ROS, LDH, ssDNA, and MDA as compared with control-untreated cells (Fig. 1B, D, F, and Fig. 2B, D). Kaempferol, in a dose-dependent manner, significantly increased ARPE-19 survival and lowered the levels of ROS, LDH, ssDNA, and MDA as compared with H2O2-treated ARPE-19 cells. The maximum alterations in cell survival and the levels of these biochemical parameters, in both cell treatments, were seen with the 2 highest doses of Kaempferol (50 and 100 μM), with no obvious significant variation in the measured parameters between both doses (Fig. 1B, D, F, and Fig. 2B, D).

Effect of Kaempferol on survival rate

Effect of Kaempferol on levels of the ssDNA
Kaempferol, in a dose-dependent manner, increases MnSOD and GSH levels in both control and H2O2-treated ARPE-19 cells
Figure 3A–D depicts the changes in levels of MnSOD and GSH in all groups of cells. H2O2 significantly lowered levels of MnSOD and GSH in ARPE-19-treated cells as compared with control-untreated cells. However, Kaempferol, in a dose-dependent manner, significantly increased levels of MnSOD and GSH in both control and H2O2-treated ARPE-19 cells as compared with control-untreated cells. However, the maximum increase in the levels of GSH and MnSOD was seen in both the control + Kaempferol and H2O2 + Kaempferol-treated ARPE-19 cells at Kaempferol doses of 50 and 100 μM. Analysis of variance has shown no significant variation in levels of GSH and MnSOD when these doses were compared with each other.
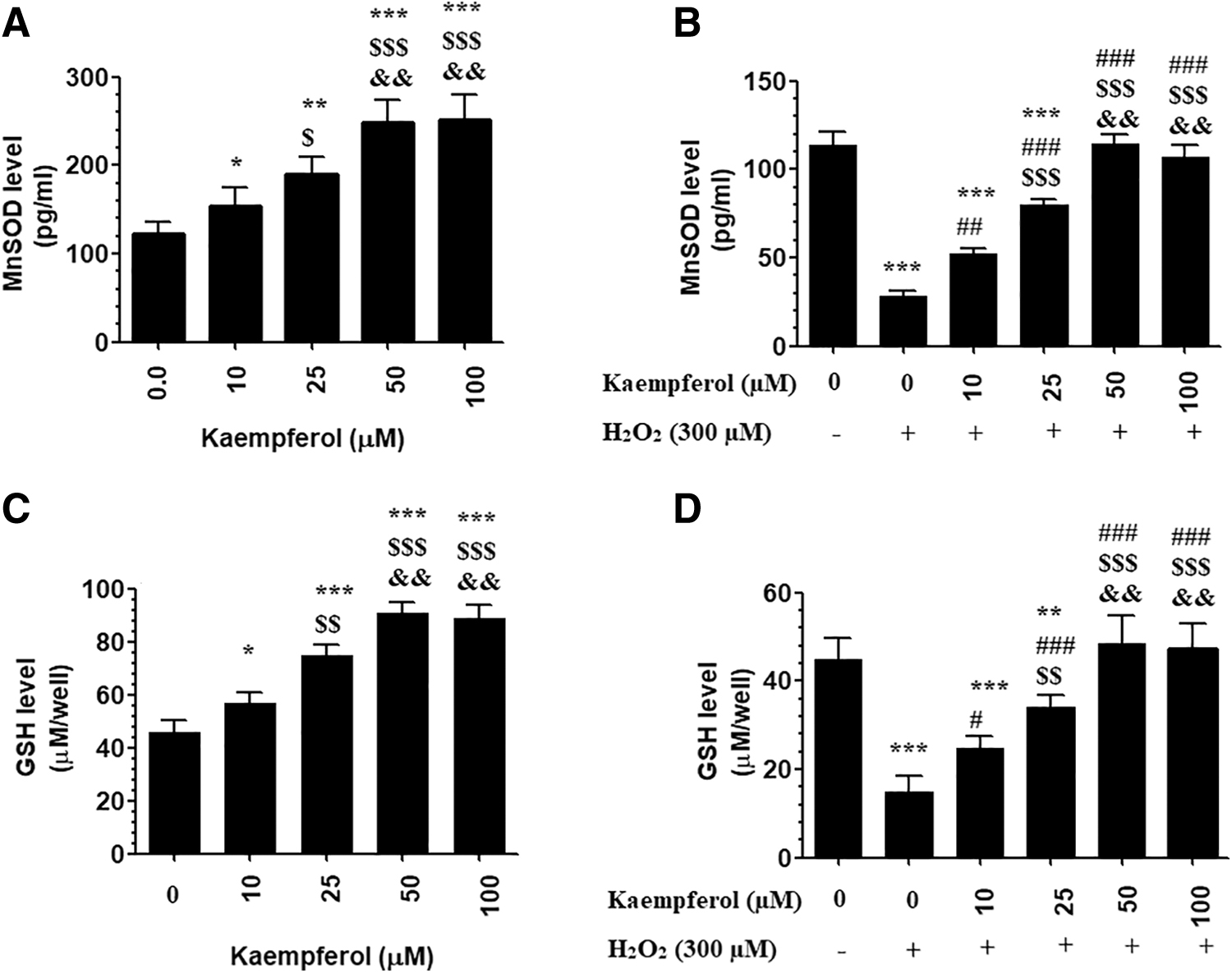
Levels of MnSOD
Kaempferol inhibits Bax, cytochrome-c, and cleaved caspase-3 in H2O2-treated cells and upregulates Bcl-2 in both control and H2O2-treated ARPE-19 cells
Total protein levels of Bax and cleaved caspase-3, as well as cytoplasmic levels of cytochrome-c, were significantly increased, but protein levels of Bcl-2 were significantly decreased in H2O2-treated ARPE-19 cells as compared with control-untreated cells (Fig. 4A–D). However, protein levels of Bax, cleaved caspase-3, and cytochrome-c were significantly decreased, whereas protein levels of Bcl-2 were significantly increased in ARPE-19 cells, which were pretreated with 50 μM Kaempferol and then incubated with H2O2 (300 μM) as compared with H2O2-treated cells (Fig. 4A–D). Of note, only protein levels of Bcl-2 were significantly increased in control + Kaempferol (50 μM)-treated ARPE-19 cells as compared with control-untreated cells (Fig. 4D).
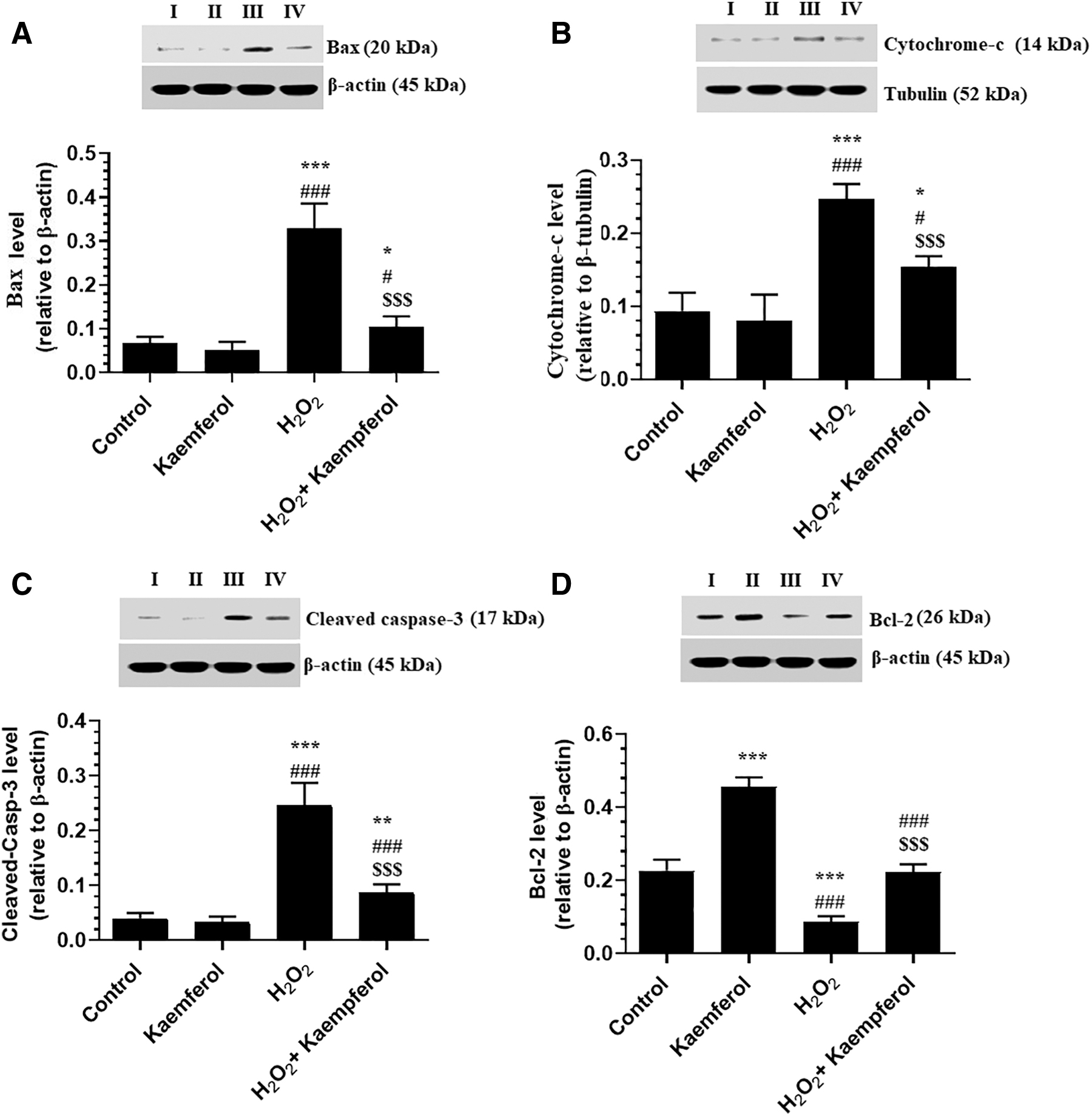
Protein levels of Bax
Kaempferol inhibits PARP1 in H2O2-treated ARPE-19 cells
Incubating the ARPE-19 cells with 50 μM Kaempferol for 24 h did not affect the nuclear activity and total, cleaved, and acetylated nuclear protein levels of PARP1 as compared with control-untreated cells (Fig. 5A–D). However, nuclear activity and total, cleaved, and acetylated nuclear protein levels of PARP1 were significantly increased in H2O2-treated ARPE-19 cells as compared with control-untreated cells (Fig. 5A–D). On the contrary, preincubating the H2O2-treated ARPE-19 cells with 50 μM Kaempferol significantly lowered the levels of all these biochemical endpoints as compared with H2O2-treated ARPE-19 cells (Fig. 5A–D).
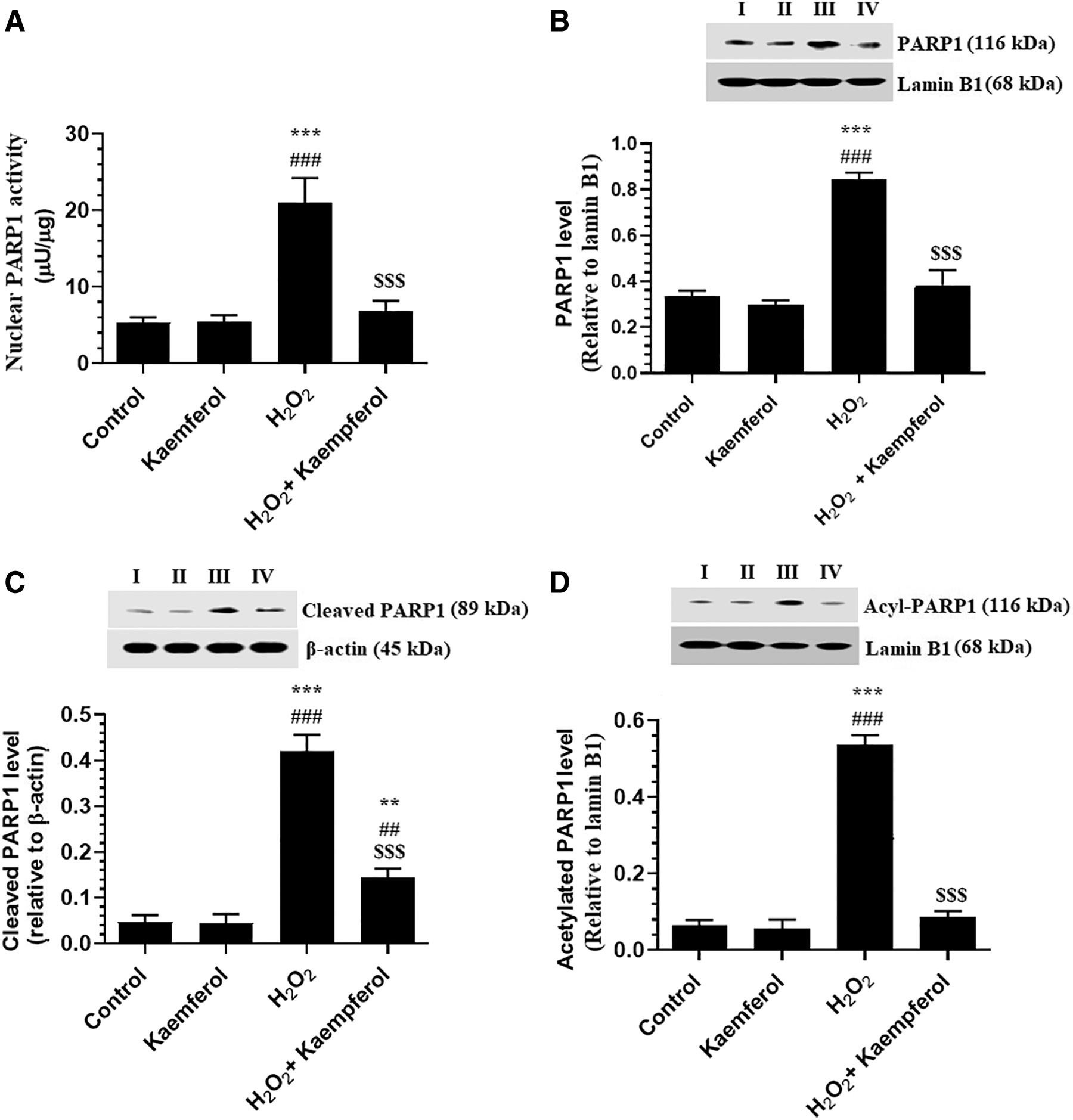
Nuclear protein levels of PARP1 activity
Kaempferol increases the nuclear protein levels of SIRT1 in both the control and H2O2-treated ARPE-19 cells
As shown in Fig. 6, nuclear protein levels and activities of SIRT1 were significantly increased, and nuclear protein levels of total FOXO-1 and acetylated-FOXO-1 were significantly decreased in control + Kaempferol (50 μM)-treated ARPE-19 cells as compared with control cells (Fig. 6A–D). On the contrary, incubating the ARPE-19 cells with 300 μM H2O2 for 24 h significantly decreased the nuclear activity and levels of SIRT1 and increased nuclear protein levels of acetylated-P53, acetylated-FOXO1, and total FOXO1 as compared with control-untreated cells (Fig. 6A–D). The alteration in these parameters was prevented when H2O2-treated ARPE-19 cells were pretreated with Kaempferol at a concentration of 50 μM (Fig. 6A–D).
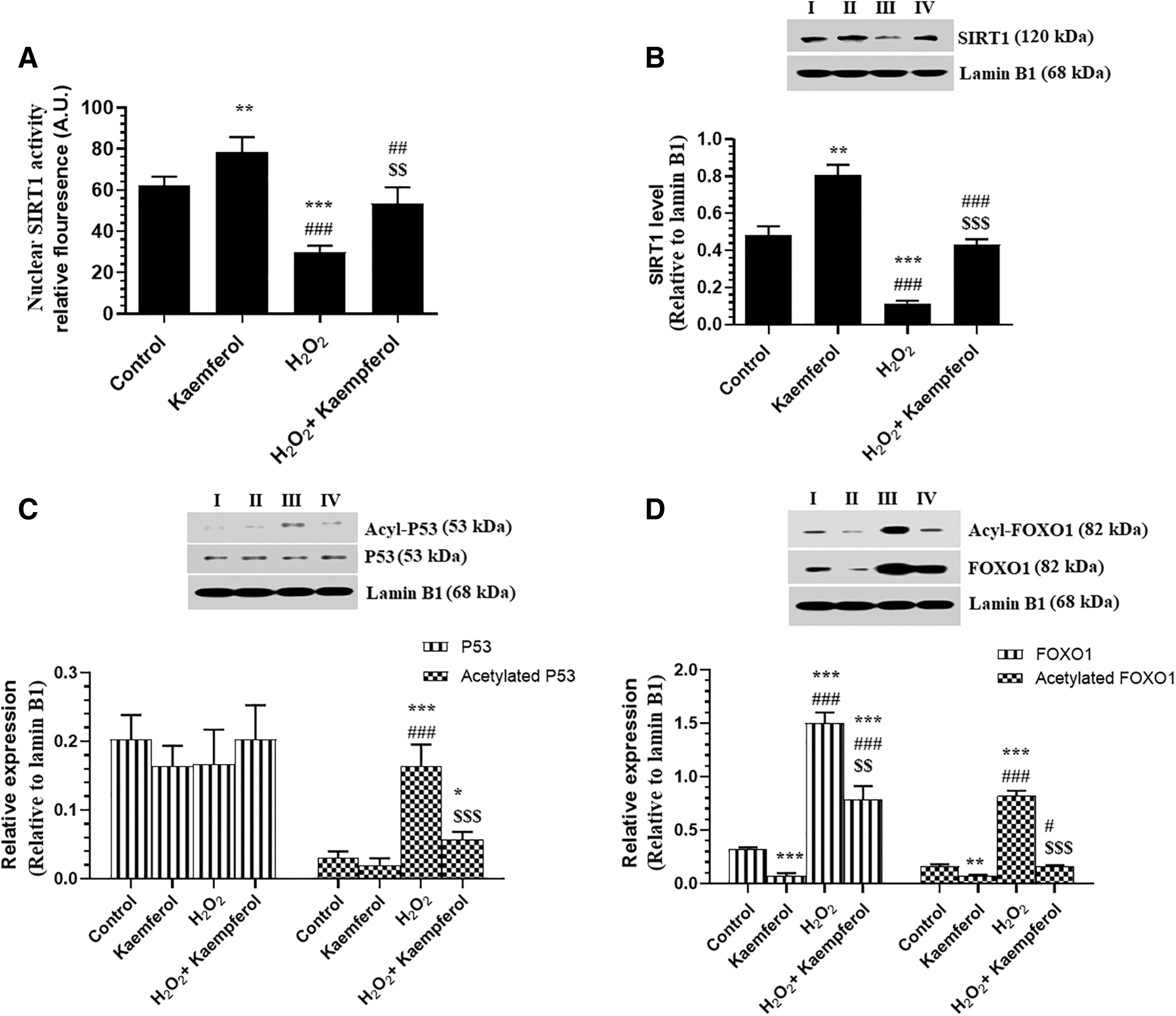
Nuclear levels of SIRT1 activity
Kaempferol decreases the nuclear accumulation and acetylation of NF-κB p6 and acetylation of Nrf2 and increases nuclear levels of Nrf2 in both control and H2O2-treated ARPE-19 cells
With no variation in protein levels of tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), a significant decrease in nuclear activity, as well as in total and acetylated nuclear protein levels of NF-κB p65, were seen in control ARPE-19 cells, which were treated with 50 μM Kaempferol as compared with control-untreated cells (Fig. 7A–D). Besides, a significant increase in total and acetylated levels of Nrf2 was seen in control + Kaempferol-treated ARPE-19 cells as compared with control-untreated cells (Fig. 8). However, protein levels of TNF-α and IL-6 and nuclear total and acetylated nuclear protein levels of NF-κB p65, as well as acetylated levels of Nrf2, were significantly increased, but total levels of Nrf2 were significantly decreased in H2O2-treated ARPE-19 cells as compared with control-untreated cells (Figs. 7A–D, 8A). On the contrary, preincubating the H2O2-treated ARPE-19 cells with 50 μM Kaempferol significantly reversed all these markers as compared with H2O2-treated ARPE-19 cells (Fig. 7A–D).
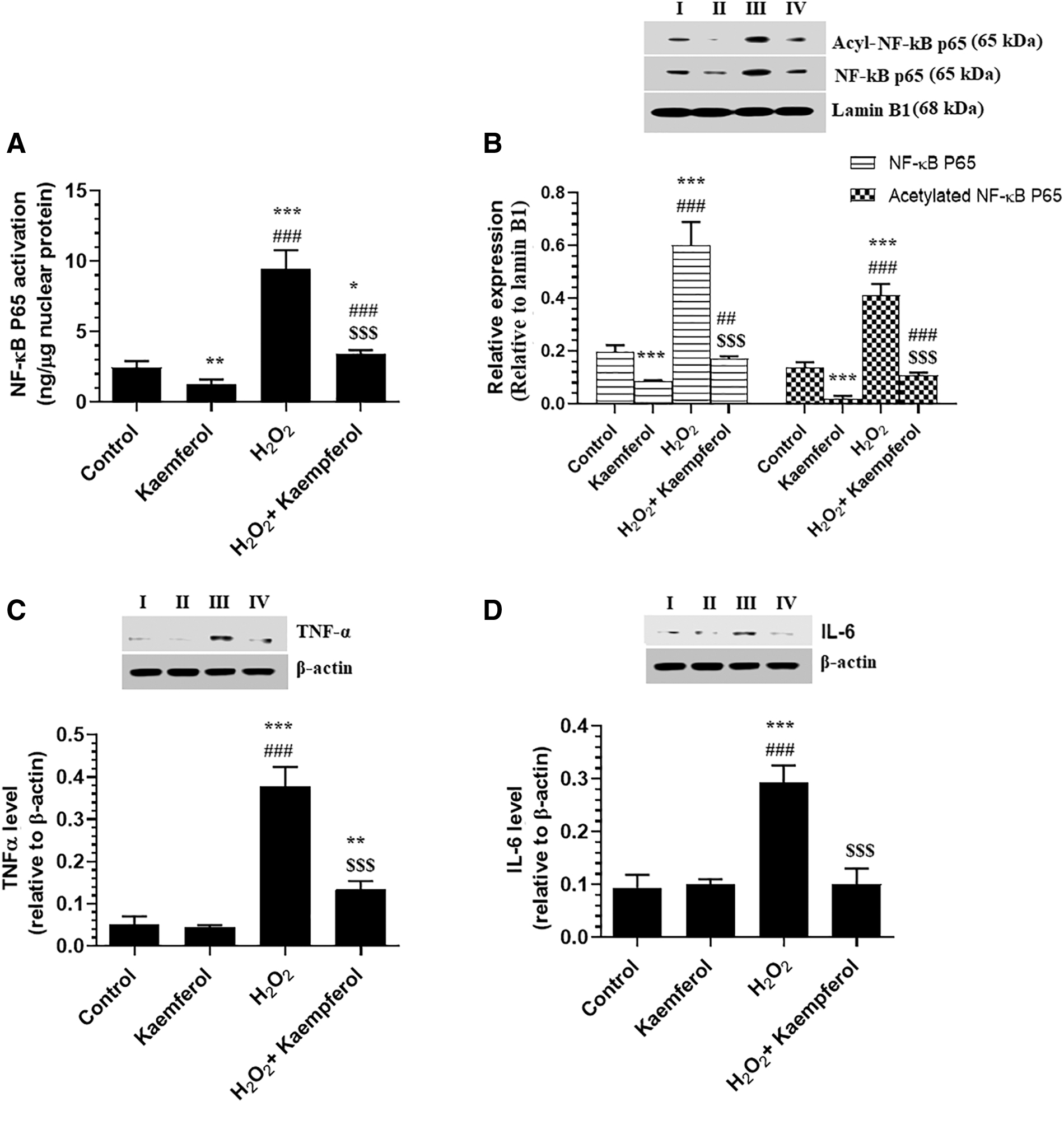
Nuclear activity of NF-κB p65, activity
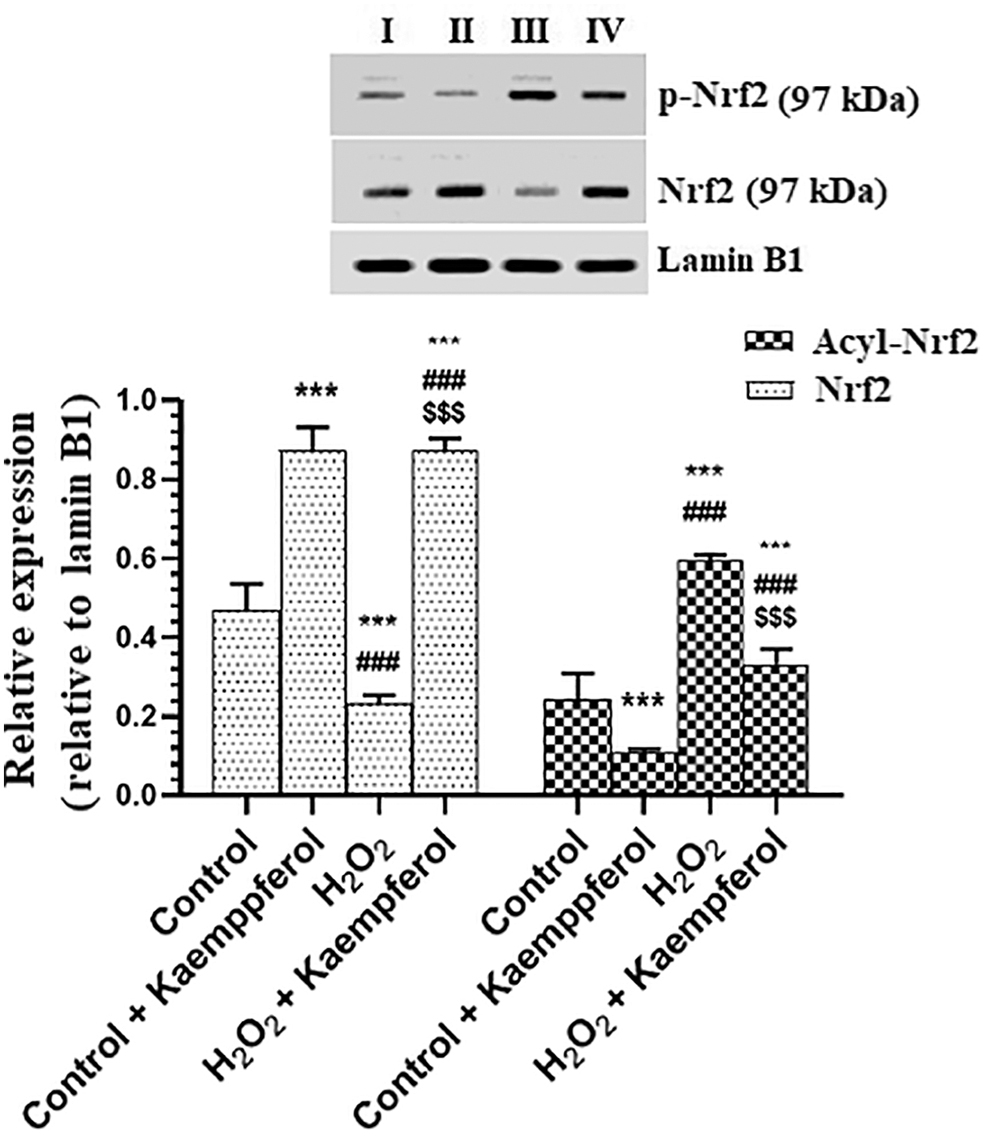
Nuclear levels of Nrf-2 and acetylated Nrf-2 in human retinal pigment epithelium cells (ARPE-19) with or without H2O2 treatment. ARPE-19 cells were cultured in DMEM/F-12 for 24 h with Kaempferol (50 μM) and then incubated for the next 24 h with 300 μM H2O2. Control cells received no treatment ***: vs. 0.0 μM at P < 0.05 and <0.001, respectively. ###: vs. Kaempferol at P < 0.001. $$$: vs. H2O2 (300 μM) at P < 0.05.
Discussion
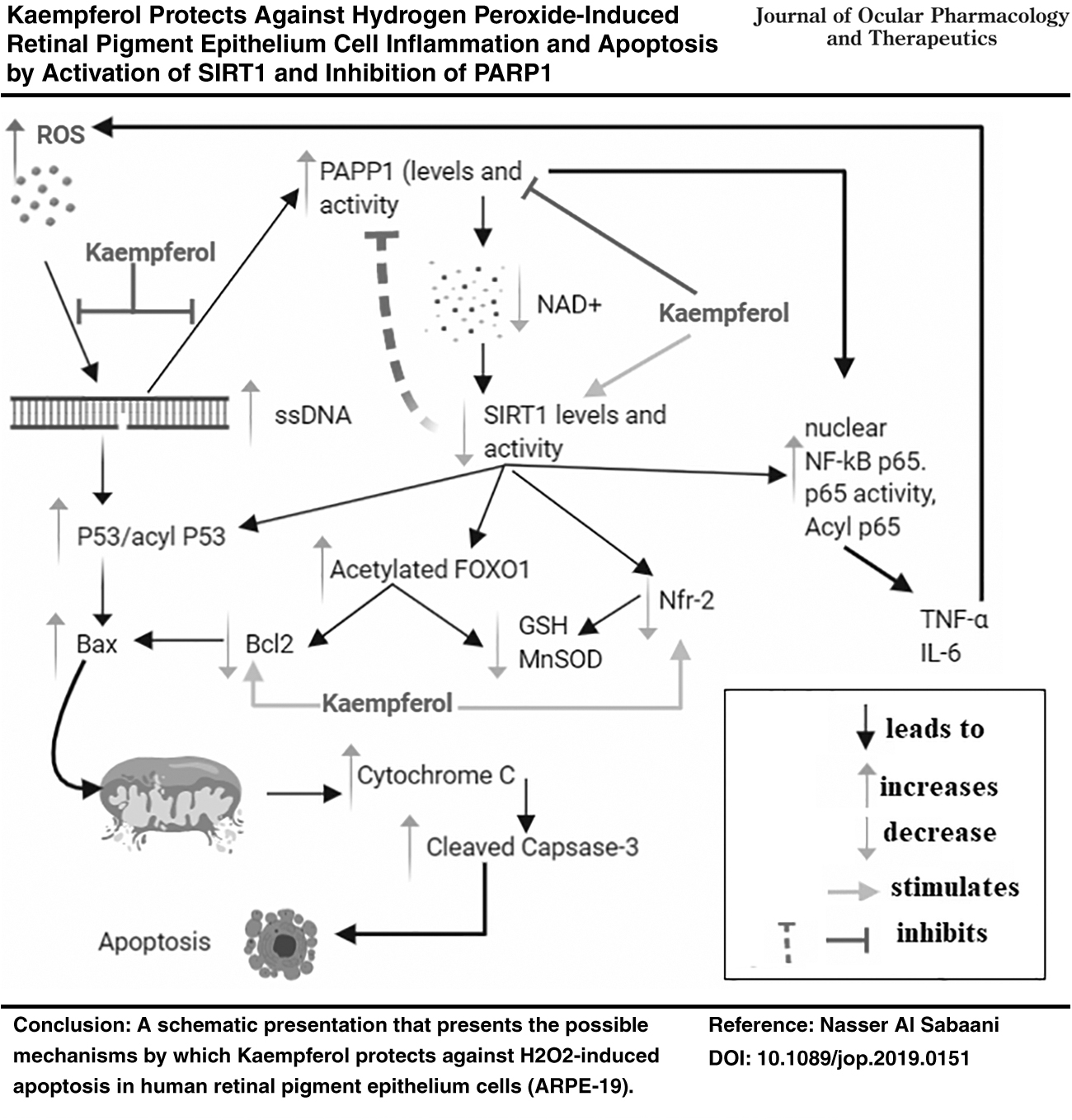
The protective effect of Kaempferol against oxidative stress (H2O2)-induced intrinsic apoptosis in RPE cells has been previously reported and was shown to be mediated by suppression of ROS, Bax, and cleaved caspase-3 and stimulation of Bcl-2 levels. 35 However, precise molecular mechanisms by which Kaempferol affords such protection are poorly described and remain enigmatic. The findings of this study extend the research within this area, and show for the first time that Kaempferol protects against (H2O2)-induced ARPE-19 cell damage by reducing the generation of ROS and inhibiting cell inflammation and mitochondria-mediated cell death of intrinsic cell apoptosis and cellular levels of inflammatory cytokines. At molecular levels, mechanism of protection afforded by Kaempferol involves, at least, (1) increasing the nuclear protein levels and the activation of SIRT1; (2) inhibition and downregulation of PARP1; (3) decreasing the nuclear levels of NF-κB p65, FOXO1; (4) deacetylation of NF-κB p65, FOXO1; (5) increasing the nuclear accumulation and deacetylation of Nrf2; and (6) upregulation of cell antioxidants, including MnSOD and GSH levels. Of interest, the stimulatory effects of Kaempferol on SIRT1 levels and deacetylation of NF-κB, p65, Nrf2, and FOXO-1, as well as on levels of MnSOD, and GSH were also shown in control ARPE-19 cells, thus suggesting its regulatory role in these signaling pathways. A schematic presentation of all these events is summarized in the graphical abstract (Fig. 8).
The significant contribution of ROS and inflammation in RPE apoptosis and subsequent loss of vision during aging or other pathological conditions is well established.7–9,42,43 In vitro, exposure of the RPE cells to exogenous H2O2 is frequently used as a model for studying the protective effect of the various compounds against oxidative stress-induced retina injury.7,42–45 Using this model, it was shown that H2O2 induces high quantities of reactive hydroxyl radicals, which initiate cell injury, inflammation, and apoptosis. 7 Also, the common modality of cell death induced by H2O2 in RPE cells is intrinsic (mitochondria-mediated), and caspase-dependent apoptosis that is associated with overproduction of ROS and inflammatory cytokines, depletion of endogenous antioxidants (GSH, SOD, catalase [CAT]), lipid peroxidation, and DNA damage.7,42,46
Similar to these findings, exposure of ARPE-19 cells of this study to 300 μM H2O2 for 24 h induced oxidative stress and inflammatory responses, increased cytoplasmic levels of cytochrome-c and total levels of cleaved caspase-3, Bax, and ssDNA and concomitantly decreased levels of MnSOD, GSH, and Bcl-2 in the treated cells. Besides, necrosis was evident in this study given the higher levels of LDH in the media of these cells. However, Kaempferol, in a dose response-dependent manner, attenuated all these adverse effects of H2O2 in ARPE-19 cells with doses of 50 and 100 μM to be the most effective doses. Hence, all the next experiments were performed using Kaempferol at a dose of 50 μM. Interestingly, we have also found a significant and dose-dependent increase in the levels of GSH, MnSOD, and Bcl-2 in control ARPE-19 cells, which were also treated with Kaempferol. These data suggest potent antioxidant and antiapoptotic effects of Kaempferol mediated by scavenging of ROS and upregulation of MnSOD, GSH, Bcl-2.
Supporting these findings, the ROS scavenging abilities and anti-inflammatory effects of Kaempferol are well reported.37,47,48 Indeed, Kaempferol inhibited lipopolysaccharides induced IL-1β and TNF-α synthesis in the activated macrophages. 49 Kaempferol also protected the neurons from oxidative damage by scavenging ROS and increasing the intracellular levels of GSH, MnSOD, and other antiapoptotic genes.50,51 Also, submicromolar concentrations of Kaempferol can scavenge numerous ROS, including superoxide, hydroxyl, and peroxynitrite radicals. 52 Besides, Kaempferol increased the expression and activity of several antioxidants such as SOD and CAT, and inhibited the activation of the xanthine oxidase, a ROS generating enzyme, in PC12 adrenal medullary cells.48,53,54 Such potent antioxidants and anti-inflammatory potentials of Kaempferol were attributed to its unique structure, which contains an oxo group at C4, double bond at C2–C3, and numerous hydroxyl groups at C3, C5, and C4.55,56 Indeed, Kaempferol can inhibit the generation of hydroxyl radicals in cells directly by chelating cuprous or ferrous ions.57,58 This could account for the significant reduction in ROS and inflammatory cytokines as demonstrated in this study. However, precise mechanisms by which Kaempferol induces upregulation of MnSOD and GSH and Bcl-2 are poorly investigated and will be our target in the next set of experiments.
The regulation of cellular antioxidants, apoptotic and antiapoptotic genes is a very complicated process, and involves numerous signaling pathways and transcriptional factors. The nuclear factor E2-related factor 2 (Nrf2) is a transcription factor that is activated in response to oxidative stress to induce the expression of several antioxidant protective proteins such as GSH, SOD, and CAT. 59 Also, Nrf2 is an important negative regulator of inflammation that inhibits NF-κB and subsequent release of the inflammatory cytokines.19,20,60,61 Interestingly, the transcriptional activity of Nrf2 can be regulated by acetylation. 62 Within this view, it was shown that activation of SIRTI not only increased the expression of Nrf2 but also increased its deacetylation to stabilize its nuclear transportation and its transcriptional activity to increase the expression of genes that encode SOD and GSH.16–18,63,64 Conversely, downregulation/inhibition of SIRT1 cellular levels significantly reduced Nrf2 protein expression and suppressed the expression of the antioxidant genes. 65 Also, SIRT1 can upregulate antioxidant and antiapoptotic genes through the deacetylation of FOXO transcription factors.15,66,67 In this regard, it was shown that nuclear accumulation of SIRT1 deacetylates FOXO-1/3 in the nucleus to increase its transcriptional activity to increase the transcription of MnSOD and CAT.66,67
Besides, it is well reported the upregulation and activation of p53 upregulates Bax and stimulates its mitochondria translocation, which in turn results in mitochondria membrane permeabilization and activates mitochondria-mediated (intrinsic) cell apoptosis. 14 On the contrary, although it has a survival role of increasing the expression of many antiapoptotic and antioxidant genes, sustained activation of NF-κB induces cell death by induction of inflammatory cytokines (TNF-α, IL-6, and IL-1β), which in turn exaggerates the production of ROS.13,69 Also, NF-κB can induce mitochondria-mediated cell apoptosis by upregulation of BCl-X(S) and Bax. 70 Furthermore, TNF-α can induce cell apoptosis by inhibiting Bcl-2 levels through the ASK1/JNK pathway. 71 As a deacetylase, SIRT1 can inhibit Bax upregulation and activation through the deacetylation of p53. 16 Also, SIRT1 can inhibit cell apoptosis and inflammation by deacetylation of NF-κB p65. 19
In this study, an interesting finding is that we also show that the pro-oxidant and apoptotic effects of H2O2 in ARPE-19 cells were associated with a reduction in total nuclear protein level and activity of SIRT1 with a concomitant increase in the total and nuclear activity of NF-κb, reduced expression of Nrf2, and increased acetylation of FOXO-1, NF-κB, Nrf2, and p53. In addition, H2O2 significantly increased protein levels of TNF-α and IL-6 in ARPE-19 cells. Supporting these findings, previous studies have shown the ability of H2O2 to reduce levels of Nrf2 in RPE cells. 44 Also, H2O2 inhibited SIRT1 levels and activities in retinal endothelial cells 72 and other cell types, including keratinocytes and bronchial cells.73,74 In the same line, H2O2 increased the nuclear activation of NF-κB and the inflammatory cytokines in retinal ganglion (RGCs) and RPE cells.57–75 Furthermore, H2O2 increased the levels of p53 in retinal cells. 78 Hence, in addition to overconsumption due to high levels of ROS, these findings suggest that H2O2 induces oxidative stress and suppresses the levels of GSH and MnSOD in ARPE-19 cells through decreasing the nuclear translocation and inhibiting SIRT1, decreasing nuclear levels of Nrf2, and increasing the nuclear accumulation of NF-κB with concurrent increase in the acetylation of FOXO-1, p53, and NF-κB.
On the contrary, the nuclear level and activity of SIRT1 and the total nuclear Nrf2 were significantly increased in both control + Kaempferol and H2O2 + Kaempferol-treated cells. These effects were also associated with a significant decrease in the nuclear levels of acetylated Nrf2. Also, control + Kaempferol or H2O2 + Kaempferol-treated ARPE-19 cells showed a decrease in the acetylation of p53 with a concurrent reduction in nuclear total and acetylated levels of NF-κB and FOXO-1. Hence, it seems logical that the stimulatory effect of Kaempferol on Bcl-2, MnSOD, and GSH, as well as on cell inflammation and survival in these cells, is a SIRT1-dependent mechanism and involves deacetylation of all these transcription factors. Given the negative cross-talk between Nrf2 and NF-κB, it could be possible that the anti-inflammatory effect of Kaempferol on ARPE-19 cells is referred to as the upregulation/activation of Nrf2. Although was poorly studies with Kaempferol, several other polyphenols and flavonoids such as piceatannol, and isoliquiritigenin, resveratrol, and quercetin enhanced cell life and inhibited cell apoptosis by stimulation and upregulation of SIRT1.79–81 Indeed, Kaempferol protected the cardiomyocytes from ischemia-reperfusion injury by upregulation and activation of SIRT1. 82 Also, Kaempferol and quercetin, at low doses (100 μM), protected against H2O2 cell apoptosis in HepG2, liver and lung cells by upregulation of Nrf2 and antioxidant genes.38,39 However, these findings are novel, and are the first to describe that the oxidant and antioxidant effects of H2O2 in ARPE-19 cells involve suppressing nuclear level and activity of SIRT1, and the protective effect of Kaempferol is mediated by contradicting these effects.
On the contrary, what causes a nuclear decrease in SIRT1 levels in H2O2-treated ARPE-19 cells and led to a reversal picture in Kaempferol-treated cells remained a challenging question. Generally, the regulation of SIRT1 in most cells remains unclear. The levels and activity of SIRT1 are mainly regulated by the cellular availability of NAD+.11–15,24 H2O2 induces DNA damage at higher concentrations, and can activate PARP1 in the retinal and nonretinal cells.83,84 However, overactivation of PARP1 can induce cell apoptosis by depletion of NAD+ and ATP and subsequent dampening of SIRT1 activity.21,24 Nonetheless, SIRT1 can also suppress the transcription and the translation of PARP1, and inhibits its activation by deacetylation.21,24
In this study, nuclear total levels, acetylated levels, and activity of PARP1 were significantly increased in H2O2-treated ARPE-19 cells, thus confirming the indispensable role of PARP1 in H2O2-induced inflammation and apoptosis in these cells. These effects indicate the existence of active DNA damage as previously shown by the higher levels of ssDNA in H2O2-treated ARPE-19 cells. However, the concomitant decrease in acetylated PARP1 could be secondary to the decreased activity of SIRT1, which normally deacetylates PARP1 to inhibit its activity. 24 PARP1 can also stimulate cell inflammation by acting as a transactivator of NF-κB p65, which could also participate in the observed increase of TNF-α and IL-6. 23 However, caspase-3 could induce PARP1 cleavage to stimulate cell apoptosis further. 85 The appearance of cleaved PARP1 is considered a signature of cell apoptosis. 85 This explains why we have also found an increase in cleaved PARP levels in H2O2-treated ARPE-19 cells. Despite these findings, if the decrease in the nuclear levels and activity of SIRT1 are caused directly by H2O2 or due to PARP1-induced reduction in NAD+ intracellular levels cannot be concluded from these data. Unfortunately, we did not measure levels of NAD+ in these cells to confirm this effect. Also, future studies at the levels of gene inhibition are required. On the contrary, and associated with reduced levels of ROS and higher nuclear levels and activities of SIRT1, Kaempferol significantly lowered levels and activities of PARP1. Hence, it could be concluded that Kaempferol protected against H2O2-induced PARP1 activation by either reducing DNA damage through scavenging ROS or directly through activating the PARP1 common inhibitor, SIRT1.
However, despite these interesting findings, this study still has some limitations. Although the ARPE-19 cells have been extensively used to study the protective effect of certain drugs against oxidative stress-induced retinal damage, some studies have shown that this cell line expresses reduced normal RPE markers such as those that regulate and maintain a barrier with strong tight junction, therefore, resembling the aged eye or an eye with pathological conditions. 86 Therefore, repeating this study in fetal primary cell lines is highly recommended to confirm our findings.
In conclusion, the findings of this study are unique to show that H2O2 induces inflammation and apoptosis in ARPE-19 cells by activation of PARP1 and concomitant inhibition of SIRT1, and the protection afforded by Kaempferol in this model implies potent stimulation of nuclear SIRT1 levels, activities, and signaling, as well as inhibition of PARP1.
Footnotes
Acknowledgments
The author thanks the technical staff at the laboratories of King Khalid University for their help in measuring some biochemical parameters of this study, and also thanks the deanship of scientific research for their continuous help.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.