
Abstract
Purpose:
To evaluate the pathological role of autophagy in dry eye diseases by detecting the autophagic degradation of RIG-I, a master RNA-sensing receptor in cells.
Methods:
RNA-sequencing analysis and qPCR analysis of the expression level of genes related to IFN-I signaling pathway was used to evaluate the inflammatory level of cells overexpressed with RIG-I or empty vector, which was further confirmed by WB analysis. Chemical treatment (3-methyladenine, chloroquine, NH4Cl, rapamycin, torin 1 or trehalose) or gene knockdown was used to modulate autophagy. When the autophagy level was regulated, the autophagic degradation of RIG-I and its pathological role in dry eye diseases were determined by detecting the protein level of RIG-I and the level of cell inflammation.
Results:
Cells that overexpressed RIG-I showed increased expression of genes involved in the IFN-I signaling pathway compared with cells transfected with an empty vector. Inhibition of autophagy leaded to the accumulation of RIG-I in HCECs, combined with the aggravation of the RIG-I-mediated IFN-I signaling pathway. Contrarily, promoting the autophagic degradation of RIG-I by trehalose treatment could alleviate IFN-I signaling pathway.
Conclusions:
Autophagy could protect the ocular surface against IFN-I signaling pathway by degrading RIG-I in HCECs. This process may restrict the overactivation of inflammation in the pathological development of dry eye disease.
Introduction
Dry eye disease (DED) is a chronic ocular surface disease caused by various factors, including aging, autoimmune diseases, diabetes, eye surgery, contact lens usage, and environmental factors. 1 Due to abnormal tear quality and dynamics, unstable tear film, or unbalanced ocular surface microenvironment, it can be accompanied by ocular surface inflammatory reaction, tissue injury, and neurological abnormalities, resulting in various ocular discomfort symptoms and/or visual dysfunction. 2
As one of the most common eye diseases, dry eye accounts for 5%–34% of the global population. Its incidence rate increases significantly with age, especially among people older than 60. 3 With the popularity of video technology, environmental pollution, and lifestyle changes, the incidence rate of dry eye has increased year by year, and the trend has been increasing. 4 DED seriously affects the quality of patients' life, but there is no radical cure at present. The research on dry eye, especially its pathogenesis and treatment scheme, has become one of the key research directions of ophthalmology.
The pathogenesis of dry eye is not completely clear. Experiments show that helper T cells on the ocular surface release immune factors and mediate noninfectious immune-related inflammatory responses, which is one of the most important pathogenic factors of dry eye. 5 In patients suffering with DED, persistent stimulation caused by excessive activation of cold fiber sensors and nociceptors, as well as tear hypertonic caused by evaporation stress, induces a transient protective adaptive response called parainflammation to restore ocular surface homeostasis. 6 While if stimulation or tissue dysfunction persists for a period of time, the parainflammation will become a persistent inflammatory state on the ocular surface. 6
The stability of tear film is destroyed, combined with increased osmotic pressure, inducing the damage of ocular surface tissue and initiating the inflammatory cascade reaction, while the immune inflammation will lead to further damage of ocular surface and develop into a periodic inflammation, aggravating the disease. 1
The dry eye model showed that inflammatory signaling pathways such as tumor necrosis factor (TNF), interferon (IFN), and the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) were activated in tear film and conjunctival epithelial cells-α, followed by the overexpressed inflammatory factors. 7 Due to the direct relationship between inflammatory factors and the pathogenesis of dry eye, reducing inflammation is an ideal therapy. Inhibitors of cellular inflammatory factors have been used as the main clinical therapy on dry eye. 8 However, the long-term use of broad-spectrum inflammatory inhibitors leads to elevated intraocular pressure and fungal infection of the cornea. 9 Therefore, it is urgent to develop newer, more specific, and less side-effect drugs to inhibit inflammatory factors.
Retinoic acid-induced gene-I (RIG-I), synthesized by gene dexd/H-box helicase 58 (ddx58), is a member of the pattern recognition receptor family. 10 By recognizing the double-stranded RNA virus, it senses intracellular virus invasion, activates interaction regulatory factor 3/7 (IRF3/7) and nuclear factor kappa-B (NF-κB) signaling pathway, and produces type I IFN and inflammatory factors, which resist virus invasion and play an antiviral role. 11 The overactivation of RIG-I will lead to the continuous release of IFN-I, which may cause self-inflammatory diseases. 12
At present, it is also reported that a single-point mutation of RIG-I will cause an autosomal dominant multisystem disease, the Singleton–Merten syndrome. Its characteristics include glaucoma, psoriasis, and so on.12,13 RIG-I plays a key role in the activation of IFN-I signaling pathway, but the relationship between RIG-I and DED has not been reported.
Autophagy, an important catabolic pathway in eukaryotes, can degrade redundant aging organelles and proteins, regulating cell component renewal and maintaining the stability of intracellular environment. 14 Autophagy also supplies energy and maintains cell survival by degrading intracellular components under stress conditions such as nutritional deficiency, DNA damage, and oxidative stress. 14 The disorder of autophagy leads to a variety of diseases, such as cellular inflammatory response, tumor, and neurodegenerative diseases.15,16 Autophagy and inflammation are 2 pivotal strategies to resist pathogens.
Encouraged evidences show that autophagy and inflammation induce and regulate each other. 16 Autophagy clears the endogenous signals that induce inflammation, such as pathogens and damaged mitochondria.17,18 Autophagy can also reduce the inflammatory response by directly degrading the components of inflammatory bodies and the downstream cytokines. 19 From the anterior segment of the cornea to the posterior segment of the retina, almost all cells express autophagy-related proteins in varying degrees, and rely on autophagy to maintain the normal structure and physiological function of cells. 20 Mutations of autophagy-related gene also directly cause eye diseases. In the mouse model of functional deletion of Beclin1 and Atg7, the key factor of autophagy, the incidence rate of glaucoma and cataract significantly increased. 21
Recently, research on the function of autophagy in dry eye has gradually increased. Eye drops containing autophagy activator rapamycin could increase vascular endothelial growth factor-A and reduce inflammatory markers (IFN-γ, IL-12) in tears of dry eye model mice, and rapamycin eye drops increase the density and area of conjunctival goblet cells and alleviate the dry eye phenotype of mice.22,23 Trehalose, another autophagy activator, could suppress the inflammatory response by upregulating autophagic flux through activating transcription factor EB (TFEB) in primary human corneal epithelial cells exposed to hyperosmotic stress, which is beneficial to cure dry eye,24,25 but the specific mechanism of autophagy regulating inflammation in the pathogenesis of dry eye has not been fully explained.
In this study, we have determined that autophagy functions as a negative inflammatory regulator in human conjunctival epithelial cells (HCECs). Mechanistically, inhibition of autophagy caused the accumulation of RIG-I, which could activate the IFN-I signaling pathway. On the contrary, activation of autophagy promoted the degradation of RIG-I and reduced the activation of IFN-I signaling pathway. In conclusion, autophagy can ameliorate the inflammation of HCECs by degrading RIG-I, which may explain the function of autophagy in dry eye.
Methods
Cell culture and treatment
HCEC line cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 (Gibco) supplemented with 6% fetal bovine serum (Gibco), 7 μg/mL insulin (Sigma–Aldrich), and 7 ng/mL epithelium growth factor (Gibco) in a 37°C incubator with a humidified 5% CO2 atmosphere. HEK293T cells were grown in DMEM (Gibco) supplemented with 10% FBS in a 37°C incubator with a humidified 5% CO2 atmosphere.
Unless otherwise stated, the chemicals were used as follows: torin1 (HY-13003; MCE), 250 nM, 4 h; chloroquine (HY-17589A; MCE), 50 μM, 4 h; 3-MA (HY-19312; MCE), 5 mM, 4 h; NH4Cl (326372; Sigma), 2 mM, 4 h; rapamycin (AY-22989; MCE), 100 nM, 4 h; and trehalose dihydrate (HY-N1132A; MCE), 100 mM, 4 h.
Antibodies
Antibodies to p-STAT1 (9167), STAT1 (14994), IFIT3 (87781S), and TFEB (37785) were from Cell Signaling Technology; antibodies to p62 (18420-1-AP), Atg7 (10088-2-AP), and Myc (16286-1-AP) were from Proteintech; and antibodies to β-Actin (A5441) and LC3 (L8918) were from Sigma.
Transfection
Transient transfection of DNA or Poly(I:C) (1 mg/mL) in cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were analyzed 18–24 h after transfection.
RNA interference
HCECs were transfected with nontargeting negative control siRNA or siRNA targeting TFEB or ATG7 by Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. Cells were analyzed 48 h after transfection. The targeted sequence is as follows:
ATG7 (AUCAGUGGAUCUAAAUCUCAATT);
TFEB (AAACGGAGCCUACUGAACA).
RNA sequencing
One microgram of RNA was used for library preparation. According to the manufacturer's recommendations, the NEBNext ultra RNA library for Illumina (NEB) was used to prepare the kit, generate the library, and add the index code to the attribute sequence of each sample. The library quality was evaluated on the Agilent Bioanalyzer 2100 system. The library was sequenced on Illumina NovaSeq and a 150 BP paired-end reading was generated. The human reference genome (GRCh38) was sequenced and read using hisat2. FeatureCounts is used to calculate the number of reads mapped to each gene. Differential expression analysis was performed using deseq2 R software package.
RNA extraction and quantitative polymerase chain reaction
Total RNA was extracted from cells using TRIzol (Invitrogen). cDNA was reverse transcribed using M-MLV reverse transcription reagents (Promega). Quantitative polymerase chain reaction (qPCR) was performed with SYBR Premix Ex Taq on a 7300 Real Time PCR System. Gene expression levels were calculated according to the 2−DDCt method and normalized against β-actin. The primer sequences are as follows:
IRF7 F: CCACGCTATACCATCTACCTGG;
IRF7 R: GCTGCTATCCAGGGAAGACACA.
ISG15 F: CTCTGAGCATCCTGGTGAGGAA;
ISF15 R: AAGGTCAGCCAGAACAGGTCGT.
CXCL10 F: GGTGAGAAGAGATGTCTGAATCC;
CXCL10 R: GTCCATCCTTGGAAGCACTGCA.
IFI44L F: TGCACTGAGGCAGATGCTGCG;
IFI44L R: TCATTGCGGCACACCAGTACAG.
IFIT1 F: GCCTTGCTGAAGTGTGGAGGAA;
IFIT1 R: ATCCAGGCGATAGGCAGAGATC.
IFI27 F: CGTCCTCCATAGCAGCCAAGAT;
IFI27 R: ACCCAATGGAGCCCAGGATGAA.
RSAD2 F: CCAGTGCAACTACAAATGCGGC;
RSAD2 R: CGGTCTTGAAGAAATGGCTCTCC.
IFNB1 F: CTTGGATTCCTACAAAGAAGCAGC;
IFNB1 R: TCCTCCTTCTGGAACTGCTGCA.
ACTIN F: GGCCCGAGCCGGAGTAGCA;
ACTIN R: GATGGACGGGAACACGGCCC.
Immunoblotting
Mammalian cells were harvested and lysed in Nonidet P-40 (NP-40) lysis buffer (1% NP-40, 1 mM MgCl2, 137 mM NaCl, 20 mM Tris-HCl, pH 7.5, 1 mM CaCl2, 10% glycerol, and protease inhibitor). Proteins were denatured and resolved on sodium dodecyl sulfate– polyacrylamide gels (SDS-PAGE), and then transferred to a polyvinylidene difluoride membrane. After blocking with 5% (w/v) bovine serum albumin, the membrane was stained with the corresponding primary antibodies and secondary antibodies. Specific bands were analyzed using an Odyssey infrared imaging system.
Luciferase assays
HEK293T cells were transfected with ISRE-α, IFN-β, and NF-κB luciferase reporter plasmids, together with Myc or Myc-RIG-I for 48 h, using Lipofectamine 2000 (Invitrogen). Then, luciferase assays were performed using a Dual Luciferase Reporter Assay System (Promega) based on the protocol provided by the manufacturer.
Results
IFN-I signaling pathway is activated by RIG-I in HCECs
To assess the potential role of RIG-I in DED, we first monitored the IFN-I signaling pathway regulated by RIG-I in HCECs. Cells that overexpressed RIG-I showed an obviously increased expression of genes involved in the IFN-I signaling pathway compared with cells transfected with an empty vector (Fig. 1A). This upregulated expression pattern was further dramatically activated by Poly(I:C) stimulation (Fig. 1A), which mimics RNA virus infection and activates RIG-I. 26 qPCR confirmed the increased expression of 8 genes related to the IFN-I signaling pathway (Fig. 1B).
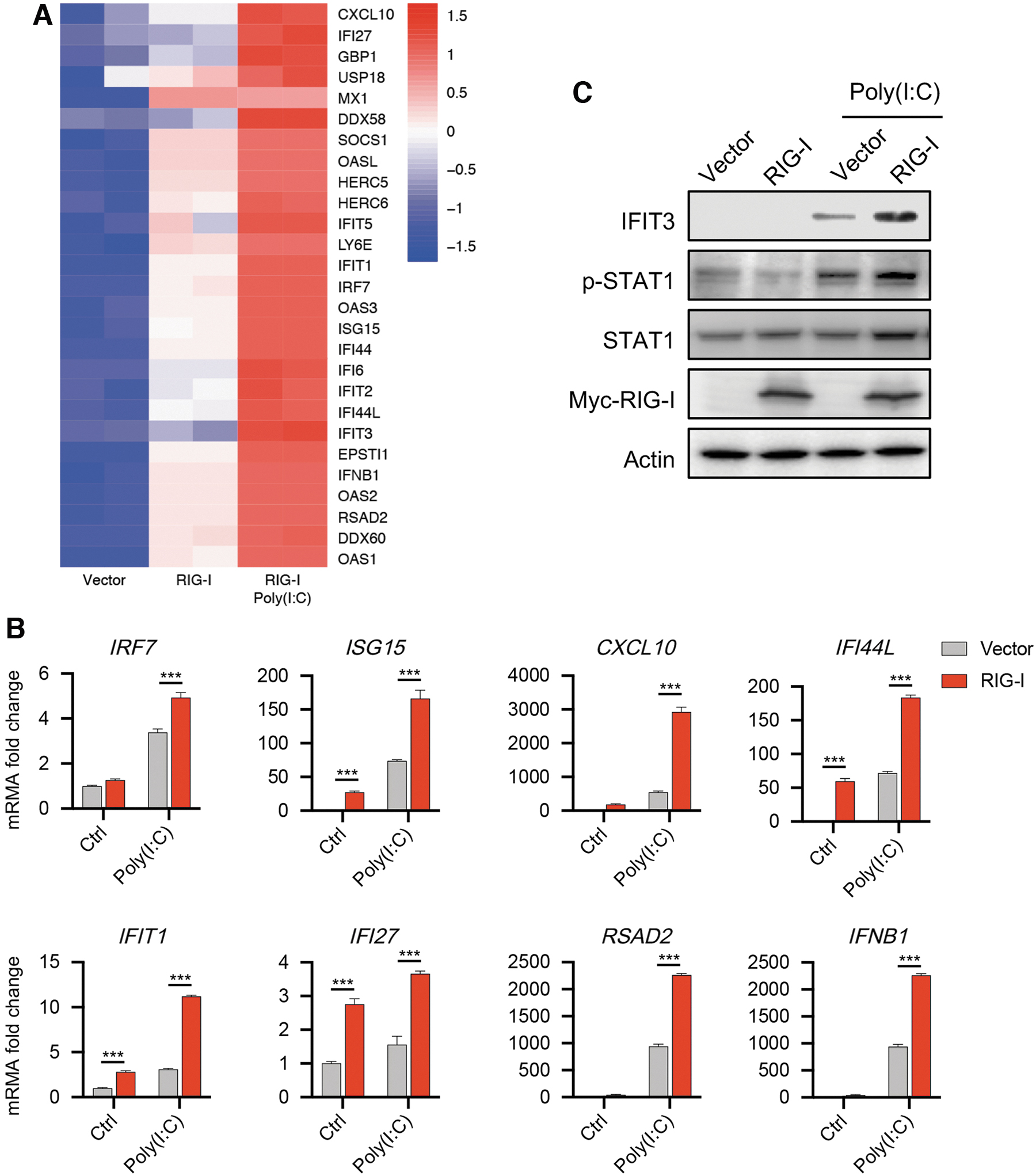
IFN-I signaling pathway is activated by RIG-I in HCECs.
Furthermore, using Western blot, we detected the higher phosphorylated level of STAT1, a key transcription activator in the IFN-I signaling pathway and the downstream protein of RIG-I, 11 which means the activation of IFN-I signaling pathway, while Poly(I:C) stimulation further improved this phosphorylated level (Fig. 1C). The protein level of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3), which acts as an inhibitor of cell migration, proliferation, signaling, and viral replication, 27 was upregulated upon RIG-I overexpression in HCECs with or without Poly(I:C) stimulation (Fig. 1C). Taken together, these data suggest that RIG-I could active the IFN-I signaling pathway in HCECs.
Inhibition of autophagy leads to the accumulation of RIG-I in HCECs
Autophagy can suppress the overactivation of IFN-I responses, maintaining immune homeostasis and avoiding damage to the host by degrading RIG-I. 28 To test the potential impact of autophagy in the regulation of IFN-I responses by degrading RIG-I, we first examined the protein level of RIG-I in HCECs. 3-methyladenine (3-mA) is a widely used inhibitor of autophagy by inhibiting phosphatidylinositol 3-kinase (PI3K). 29 When cells were treated with 3-MA, the protein level of RIG-I accumulated significantly (Fig. 2A).
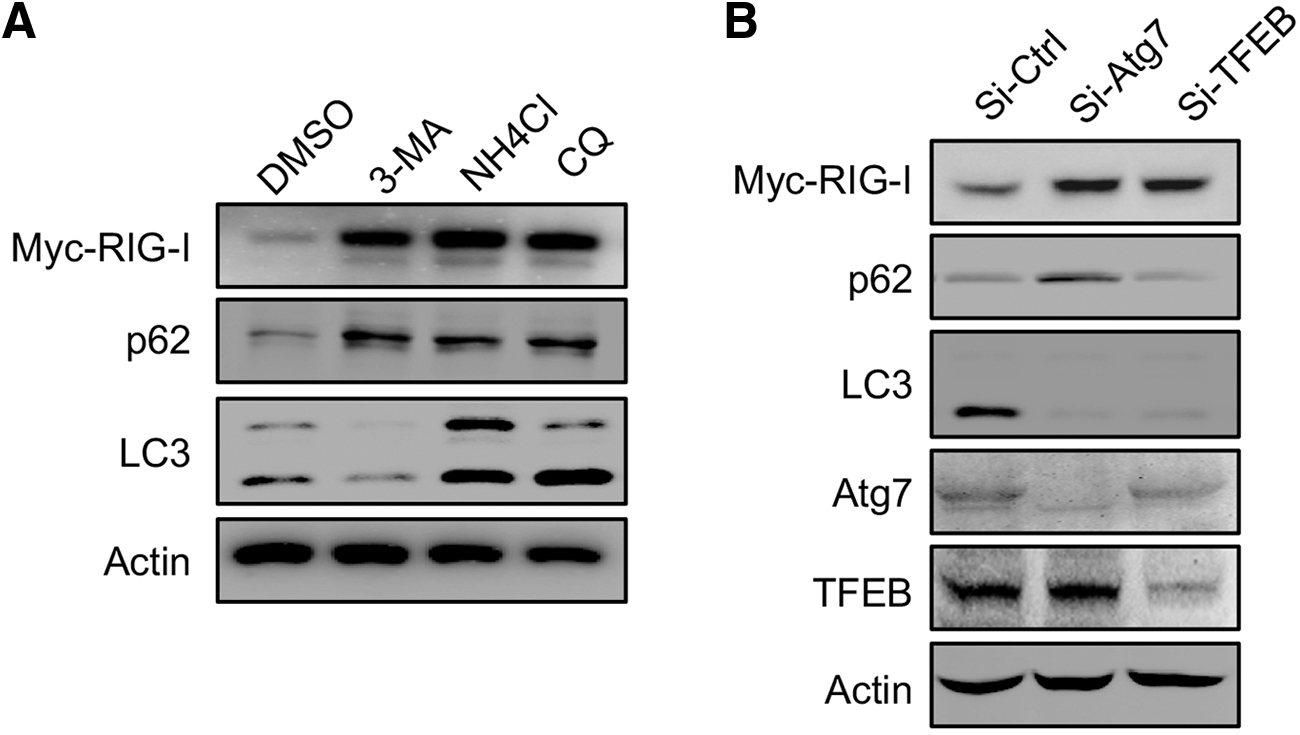
Inhibition of autophagy leads to the accumulation of RIG-I in HCECs.
The same results were obtained from cells treated with chloroquine (CQ) and NH4Cl, 2 lysosome inhibitors, blocking autophagic degradation in lysosome (Fig. 2A). 29 The significant change in the protein level of p62 and LC3-II represents the successful block of autophagy by these 3 chemicals (Fig. 2A). 29 Moreover, we used genetic methods to inhibit autophagy by knocking down Atg7, the essential protein in autophagy, 15 or knocking down TFEB, an important transcription factor in the expression of genes for autophagosome formation and lysosome production. 30 The accumulation of RIG-I was also presented by knocking down the 2 genes (Fig. 2B). Therefore, inhibition of autophagy leads to the accumulation of RIG-I in HCECs.
Inhibition of autophagy aggravates the IFN-I signaling pathway activated by RIG-I
Then, we detected the IFN-I responses in HCECs on inhibiting autophagy. Significantly, the expression of genes related to IFN-I signaling pathway was increased by treating with CQ (Fig. 3A). To further confirm this, we checked the promoter activity of IFN-β and NF-κB by using luciferase reporter assay. We found that RIG-I-WT overexpressed cells showed a higher luciferase activity than cells transfected with a vector, which is further promoted by CQ treatment (Fig. 3B). The same results were also presented using a luciferase reporter carrying the IFN-stimulated response element (ISRE) (Fig. 3B). Taken together, inhibiting autophagy may aggravate the RIG-I-mediated IFN-I signaling pathway.
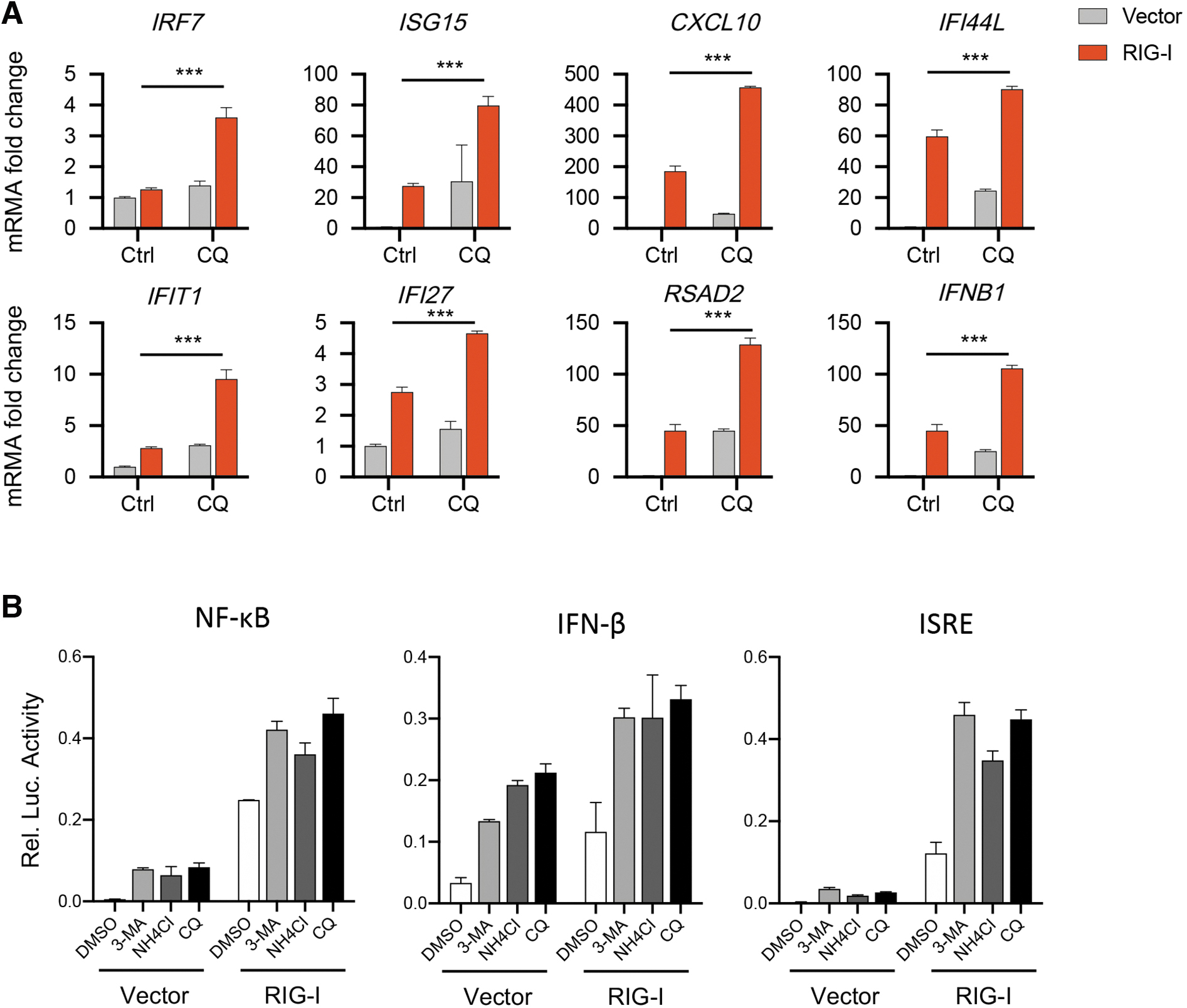
Inhibition of autophagy aggravates the IFN-I signaling pathway activated by RIG-I.
Trehalose alleviated IFN-I signaling pathway by promoting the autophagic degradation of RIG-I
We next investigated the function of autophagy activator on RIG-I and IFN-I signaling pathway. First, we treated cells with rapamycin and torin 1, 2 potent activators of autophagy by inhibiting mammalian target of rapamycin kinase. 29 Both activators promoted the autophagic degradation of RIG-I. Similar results were obtained in cells treated with trehalose, an autophagy enhancer, suppressing the inflammatory response by promoting autophagy24,25 (Fig. 4A). These results suggest that activation of autophagy can promote the degradation of RIG-I.
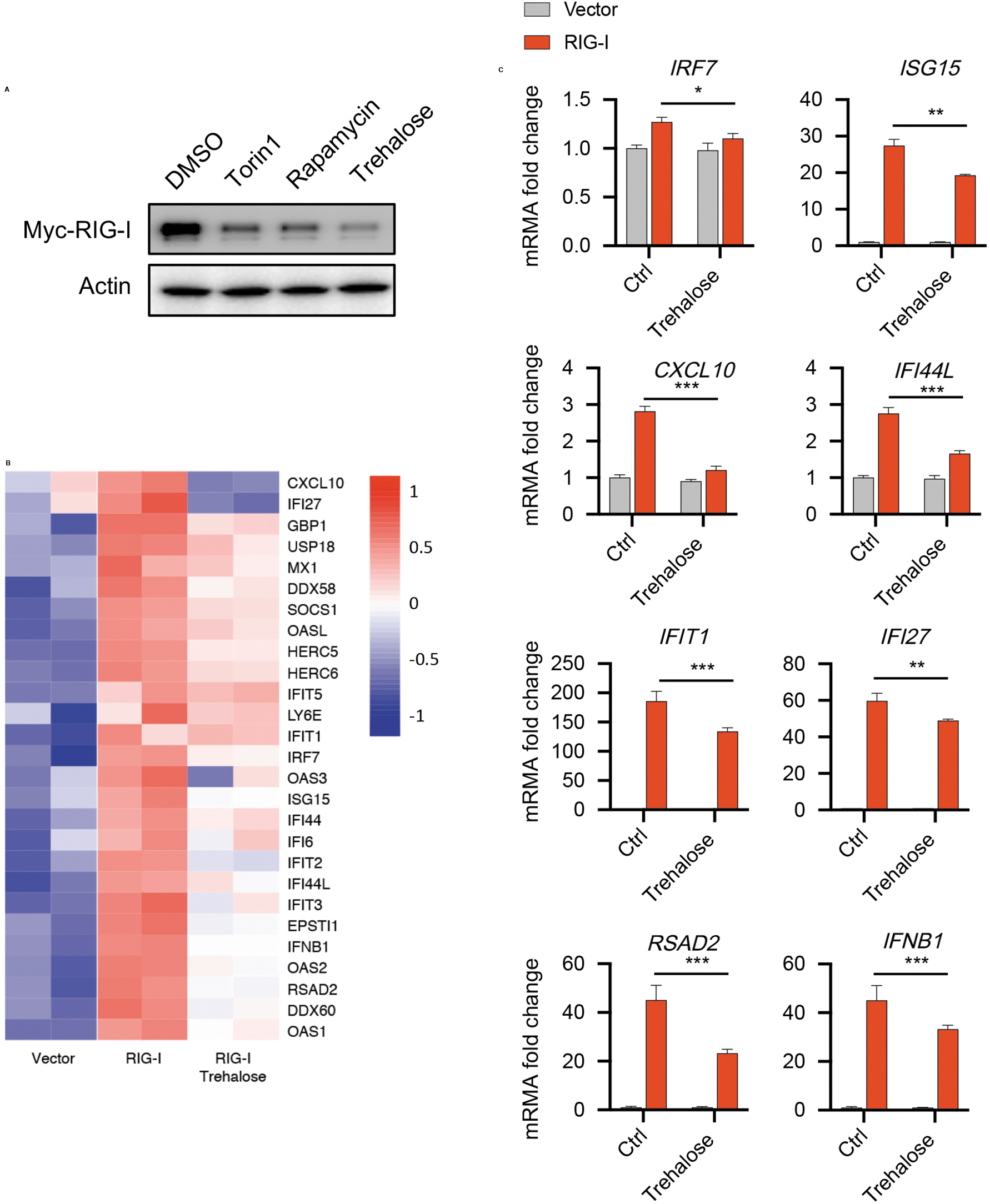
Trehalose alleviated IFN-I signaling pathway by promoting the autophagic degradation of RIG-I.
We then examined the function of trehalose in regulating IFN-I signaling pathway. The expression of gene in IFN-I signaling pathway was remarkably downregulated by trehalose stimulation (Fig. 4B), which is further confirmed by qPCR (Fig. 4C). Taken together, these data suggest that trehalose could alleviate IFN-I signaling pathway by promoting the autophagic degradation of RIG-I.
Discussion
Autophagy could alleviate the symptoms of dry eye by reducing the level of cellular inflammation, 21 but the mechanism of autophagy in this process has not been fully clarified. Here, our results demonstrate that autophagy could protect the ocular surface against IFN-I signaling pathway by degrading RIG-I, a master RNA-sensing receptor, in HCECs. This process may restrict the overactivation of inflammation in the pathological development of DED upon viral infection.
IFN-I is an important effector molecule of vertebrates against acute viral infection, which can resist viral infection by activating the JAK/STAT signal pathway and transcribing a series of interferon-induced genes. 31 The activity of IFN-I signal is closely regulated and maintains self-homeostasis through an autocrine loop. Insufficient or missing IFN leads to the defect of antiviral immune function, while an abnormal continuous activation will lead to the increase of inflammation level. 32 In the conjunctival epithelium, long-term stimulation of IFN will lead to squamous metaplasia and goblet cell secretion dysfunction and loss. 33 With the decrease of mucus secreted by goblet cells, the lipid layer becomes thinner, which further destroys the stability of tear film and leads to DED.
In our results, overexpression of RIG-I can activate IFN-I signaling pathway in HCECs, and the IFN-I response is further improved after stimulation of mimic RNA virus. This result proves for the first time that RIG-I regulates IFN-I signaling pathway in HCECs, and also provides a new target for the treatment of diseases caused by ocular surface excessive inflammation, including DED.
In recent years, autophagy has been considered a very promising target for the treatment of DED, and several drugs have successfully alleviated ocular surface inflammation and dry eye symptoms by promoting autophagy. However, the specific mechanism of this process is still unclear. We treated cells with a variety of autophagy activators and inhibitors, finding that autophagy can negatively regulate the protein level of RIG-I, accompanying with the downregulation of IFN-I inflammatory pathway in HCECs. This not only confirms the previous study on the autophagic degradation of RIG-I, 28 but also suggests that autophagy may prevent the occurrence of DED by directly degrading RIG-I and avoiding excessive activation of IFN-I inflammatory pathway in HCECs.
Trehalose is a natural nonreducing disaccharide. Due to its nontoxic side effects, it attracted rising interest in the development of various applications in the food, cosmetics, and pharmaceutical industries. 34 The evidence collected from randomized-controlled trials and preliminary studies in human subjects strongly suggests that trehalose can directly improve the condition of the affected eye by blocking the “vicious circle” of dry eye at multiple points, which will help to achieve healthy tear film homeostasis and minimize the chance of returning to a bad state.35,36 Although the mechanism is unclear, through different pathways, trehalose can activate autophagy in different cell type and tissues, including the cornea. 35 We found that trehalose could activate autophagy in HCECs, similarly to the 2 commonly used autophagy activators rapamycin and torin1. Trehalose promotes the degradation of RIG-I and the inhibition of the corresponding IFN-I signaling pathway by activating autophagy.
These results raise a new possible mechanism of trehalose in alleviating dry eye and ocular surface inflammation, and also provide more support for the clinical application of commercially available eye drops containing trehalose.
Conclusions
RIG-I activates IFN-I signaling pathway in HCECs. Inhibition of autophagy leads to the accumulation of RIG-I, which further promotes the sustained activation of IFN-I signaling, While trehalose treatment could alleviate IFN-I signaling by accelerating the autophagic degradation of RIG-I.
Footnotes
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
All the costs of the experiments are provided by the Taizhou Hospital of Zhejiang Province. The authors had complete and sole independent control over the design of the research protocol, data collection and analysis, and drafting and finalizing the article.