
Abstract
Purpose:
This study aimed to investigate the therapeutic potential of AZD6738, an ataxia-telangiectasia and rad3-related (ATR) kinase inhibitor, in preventing corneal neovascularization (CNV) by exploring its effects on autophagy regulation and angiogenesis.
Methods:
Human umbilical vein endothelial cells were cultured and treated with varying concentrations of AZD6738 and vascular endothelial growth factor (VEGF) to assess cell viability, migration, and tube formation. A corneal alkali burn model in Sprague-Dawley rats was established to evaluate the in vivo effects of AZD6738 on CNV. Autophagy was assessed using monodansylcadaverine (MDC) staining, western blotting, and qRT-PCR to measure the expression of autophagy-related markers and key proteins involved in the PI3K-AKT pathway. Immunohistochemistry and immunofluorescence staining were employed to examine histological changes and the expression of markers related to neovascularization and fibrosis.
Results:
The study demonstrated that AZD6738 significantly inhibited cell viability in a dose-dependent manner. AZD6738 effectively reduced VEGF-induced cell migration and tube formation. Moreover, the introduction of AZD6738 enhanced autophagy, as indicated by increased MDC staining, upregulated Beclin1 expression, and an elevated LC3 II/I ratio. The inhibitor also suppressed the PI3K-AKT pathway, reducing VEGF and VEGFR2 expression, and decreasing the phosphorylation levels of AMPK and AKT. In an experimental CNV model, AZD6738 treatment resulted in a significant reduction in CNV, with fewer and shorter blood vessels observed, as well as changes in autophagy-related proteins.
Conclusions:
AZD6738 showed potential in preventing CNV. Its ability to enhance autophagy and inhibit PI3K-AKT-VEGF pathways in angiogenesis suggests that AZD6738 could be an effective treatment strategy for CNV.
Introduction
The cornea is a transparent and avascular tissue crucial for maintaining the eye’s refractive function. Various pathological conditions, including infection, trauma, and ocular surface surgeries, can disrupt the balance between pro-angiogenic and antiangiogenic factors, leading to corneal neovascularization (CNV). CNV is a major cause of blindness worldwide, as newly formed vessels can obstruct light, impair vision, and negatively impact the outcomes of penetrating keratoplasty.1–3
Despite the existence of several treatment options for CNV, including anti-vascular endothelial growth factor (anti-VEGF) therapy, steroids, and surgical interventions, their effectiveness is often limited and can be accompanied by undesirable side effects.4,5 This underscores the urgent need to explore novel therapeutic approaches that target the underlying mechanisms of CNV while minimizing adverse effects.
Recent studies have highlighted the critical role of autophagy, a cellular process responsible for degrading and recycling damaged cellular components, in maintaining corneal homeostasis. 5 In CNV, autophagy has been shown to influence the inflammatory response and the polarization of macrophages, which are key players in angiogenesis.6–9 Enhancing autophagy using agents such as rapamycin has demonstrated potential in mitigating CNV, suggesting that modulating autophagy could be a promising strategy for treating CNV and other inflammation-related ocular disorders.10,11 The PI3K/AKT-mTOR pathway is a central regulator of autophagy, in which its activation leads to the suppression of autophagic, and conversely, its inhibition results in the induction of autophagy. This interplay is crucial for cellular responses to stress and has significant implications in various diseases, including corneal disorders.7,12
In this context, we turned our attention to AZD6738 (ceralasertib), a potent ATR kinase inhibitor known for its ability to induce cell death and senescence in various cancer cell lines, including non-small cell lung and human breast cancer cells.13–15 ATR kinase inhibitors have been shown to inhibit hypoxia inducible factor 1 (HIF-1) induced VEGF production, a key factor in angiogenesis. 16 Additionally, AZD6738 has been reported to reduce conjunctival fibroblast activation via the CHK1/p53 and PI3K/AKT pathways, thereby decreasing tissue fibrosis and lowering intraocular pressure.17,18 Notably, ATR inhibition has been associated with enhanced autophagy,19–21 making AZD6738 an intriguing candidate for further exploration in corneal diseases.
Our study aimed to investigate the therapeutic potential of AZD6738 in preventing CNV, particularly by examining its effects on autophagy regulation and angiogenesis. By elucidating the underlying mechanisms, we sought to enhance the understanding of ATR inhibitors’ role in managing CNV and contribute to the development of more effective treatments for this sight-threatening condition.
Methods
Materials
AZD6738 (No. HY-19323, 10 mM in dimethyl sulfoxide) and VEGF (No. HY-P7110A) were purchased from MedChemExpress (China). The dimethyl sulfoxide (DMSO) used in our study was purchased from Solarbio Science and Technology Co. (No. D8371), and it was used as a solvent for the drugs administered in the study. Antibodies against p-AMPK (No. 2535S) and AMPK (No. 5831S) were acquired from Cell Signaling Technologies (USA). Antibodies against mTOR (No. AP24743), VEGF (No. AP23759), and p62/SQSTM1 (No. A19700) were obtained from Abclonal (China). Antibodies against p-AKT (No. T40067F), AKT (No. T55561F), and LC3 (No.T55992S) were obtained from Abmar (China), Beclin1 (No. 11306-1-AP), VEGFR2 (No. 26415-1-AP), β-actin (No. 81115-1-RR), GAPDH (No. 60004-1-Ig), Goat anti-rabbit IgG (H + L; No. SA00001-2), and Goat anti-mouse IgG (H + L; No. SA00001-1) were obtained from Proteintech (China). Chloroquine (CQ, No. T8689) and Rapamycin (RAPA, No. T1537) were purchased from TargetMol (China).
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were obtained from Service Biotechnology Co. (Wuhan, China). HUVECs were incubated in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum. The cells were then placed in a 5% CO2 atmosphere at 37°C. Subsequent experiments utilized cells at 90% confluency. The cells were divided into the following groups: the control, VEGF (10 μg/mL) treated, VEGF + AZD6738 treated (1 or 5 μM), VEGF + AZD6738 + CQ (10 μM) treated groups, VEGF + CQ (10 μM), and VEGF + RAPA (20 μM). For VEGF + CQ/RAPA groups, CQ/RAPA was given for 24 h. In VEGF + AZD6738 + CQ/RAPA groups, cells were stimulated with VEGF for 24 h, treated with AZD6738 for 48 h, and CQ/RAPA was added in the last 24 h.10,22
Cell proliferation assay
Cell viability to evaluate the cytotoxicity of AZD6738 on HUVEC was assessed by the use of a Cell Counting Assay Kit-8 (Abbkine Scientific Co., Ltd., China), according to the manufacturer’s instructions. HUVECs were placed in 96-well plates at a density of 5 × 103 cells per well overnight. The cells were then exposed to varying concentrations (0.25/0.5/1/2/5/10 μM) of AZD6738 for 24 and 48 h, respectively. Following treatment, the cells were rinsed with PBS, and subsequently, 100 μL of CCK-8 in serum-free DMEM was added to each well. The cells were then incubated for 1 h in the dark, and the optical density at 450 nm was measured using a microplate reader (Victor Nivo, PerkinElmer, USA).
Cell migration assay
HUVECs were harvested at a concentration of 1.5 × 105 cells/mL and plated onto 6-well dishes. After reaching full confluence, a vertical scratch was created using a 200 μL pipette tip, followed by 3 washes with PBS. Subsequently, the cells were exposed to varying concentrations of AZD6738 (1 or 5 μM) for 0, 24, and 48 h, and images were captured via an optical microscope (TH4-200, Olympus, Japan). Cell migration was quantified using ImageJ software with the formula: Cell mobility = (initial wound area − final wound area)/initial wound area × 100%.
Cell tube formation assay
Matrigel was transferred to a precooled 96-well plate with 50 μL per well and then polymerized at 37.0°C for 30 min. HUVECs treated with AZD6738 at 1 or 5 μM concentrations were introduced into the Matrigel for 8 h. Images were captured using the optical microscope (TH4-200, Olympus, Japan) and calculated using ImageJ software.
EdU cell proliferation assay
Cells were seeded in 96-well plates at 5 × 103 cells per well and treated with different groups (Control, VEGF, AZD-5, RAPA, CQ, AZD-5 + RAPA, and AZD-5 + CQ) for 24 h. EdU (Suzhou Ue Biotech Co., C6045M) was then added and incubated for 2 h to label proliferating cells. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and stained with 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/mL) to mark nuclei. Fluorescence microscopy was used to capture images of EdU (red) and DAPI (blue) distribution. The proportion of EdU-positive cells was quantified.
Transwell migration assay
For each Transwell migration assay, the upper chamber was filled with complete media, either supplemented with or without AZD6738, under normal conditions after the HUVECs (5 × 104 cells) had been starved overnight. The lower chamber was filled with 500 μL of DMEM medium containing 10% fetal bovine serum. After a 24-h incubation, the cells were fixed in 4% paraformaldehyde for 15 min. They were then stained with crystal violet and counted using an optical microscope (TH4-200, Olympus, Japan).
Monodansylcadaverine staining
Monodansylcadaverine (MDC) staining was performed to evaluate autophagy. HUVECs were plated in two 24-well dishes and harvested following exposure to 1 or 5 μM AZD6738 and 10 ng/mL VEGF for 48 h. Subsequently, 0.05 mmol/L MDC (Solarbio Science and Technology Co., Ltd., Beijing, China) was added to the wash buffer and incubated at 37°C for 30 min. Subsequent observation was performed under a fluorescence microscope in darkness on an anti-fluorescence quenching slide.
Western blot analysis
HUVECs seeded in two 6-well plates were collected after being treated with AZD6738 (1 or 5 μM) and 10 ng/mL VEGF for 48 h. Total proteins were extracted from HUVECs using radioimmunoprecipitation assay lysis buffer (Solarbio Science and Technology Co., Ltd., Beijing, China) containing 1% phenylmethanesulfonyl fluoride (PMSF; Solarbio Science and Technology Co., Ltd., Beijing, China). The proteins were then prepared for analysis by SDS-PAGE using 8%–12% polyacrylamide gel and transferred onto a polyvinylidene fluoride membrane (Millipore, Burlington, MA, USA). After blocking in 1% bovine serum albumin (BSA) for 1 h, the membranes were incubated with primary antibodies mTOR, VEGF, VEGFR2, p62, AKT, p-AKT, Beclin1, LC3 overnight at 4°C. After washing with PBS 3 times, the membranes were exposed to secondary antibodies. Results were visualized using the enhanced chemiluminescent luminol (ECL) detection kit and analyzed using ImageJ.
RNA isolation and quantitative real-time RT-PCR
Total RNA was extracted from cultured HUVECs or corneal tissues using commercial kits (Aidlab, Beijing, China) following the manufacturer’s instructions. Reverse transcription was carried out using a kit from TaKaRa (Otsu, Japan). Quantitative real-time RT-PCR (qRT-PCR) was performed with SYBR Green Real-Time PCR Master Mix (TaKaRa, Otsu, Japan). The amplification program included an initial denaturation step at 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, 57°C for 30 s, and 75°C for 10 s; then, the melting curve analysis was conducted at once from 65°C to 95°C. Transcript abundance was normalized to GAPDH levels and calculated using the 2−ΔΔCt method. The primers are listed in Supplementary Table S1. The results of the relative quantitative real-time PCR were analyzed by the comparative threshold cycle method and normalized to GAPDH as the reference gene.
Animals and drug administration
Sixty male Sprague-Dawley rats (6–8 weeks, 180–220 g), with healthy corneas and no evidence of preexisting ocular diseases, were obtained from the Laboratory Animal Center at Nanchang University. All studies were performed following approval from the Nanchang University Ethics Committee (Approval No. NCULAE-20221031171, dated October 31, 2022) and in accordance with the Association for Research in Vision and Ophthalmology. Animals were given free access to standard rodent chow and water and kept in a standard pathogen-free environment at 25°C ± 1°C, relative humidity 55% ± 5%, and alternating 12-h light–dark cycles (lights on at 8:00 and lights off at 20:00).
Corneal alkali burn
To induce the corneal alkali burn model, rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg). A topical application of 0.5% proparacaine hydrochloride ophthalmic solution was administered to the right eye, before the application of a 3.00 mm diameter circular filter paper soaked in 1 M NaOH on the cornea for 30 s. The cornea was then washed with 20 mL sterile balanced salt solution (0.9% NaCl, Alcon Laboratories). Subsequently, ofloxacin ophthalmic drops (Zhongshan Wanhan Pharmaceutical Co., Ltd., China) was administered topically at 30 min post-injury. Each rat had its right eye subjected to alkali burn injury, while the left eye served as a normal control.23,24 The rats were administered either vehicle (1% DMSO in PBS, n = 30) or AZD6738 (100 μM AZD6738 with 1% DMSO in PBS, n = 30) eye drops 4 times daily from 8:00 to 20:00, with each administration spaced 3 h apart, for 14 days post-alkali burns.
Slit-lamp evaluation, corneal opacity assessment, and measurement of corneal neovascularization
The opacity and neovascularization in rats were assessed using a slit-lamp microscope (Chongqing Sunkingdom Medical Instrument Co., Ltd., China). The degree of opacity was rated on a scale from 0 to 4, where 0 represented a completely clear cornea and 4 signified total opacity.25,26 Neovascularization was measured according to Robert’s model formula: S = C/12 × 3.1416 × [r2 − (r − I)2], with C representing the duration of neovascularization accumulation around the cornea, r as the corneal radius, and I as the distance from the corneal edge to the neovascularization location. 27
Hematoxylin–eosin staining
Rats from the alkali burn model and the AZD6738 treatment groups were euthanized on day 14 post-injury. Their eyeballs were dissected and fixed with 4% paraformaldehyde overnight, embedded in paraffin wax, sliced into 5 μm sections, and stained with hematoxylin for 1 min and eosin for 2 min. The histopathological findings, which included the intensity of neovascularization, inflammatory cell infiltration, and fibroblast activity, were evaluated using a modified version of the scoring system previously defined by Ozdemir et al. 28
Immunohistochemical staining
Sections were rehydrated and endogenous peroxidase activity was blocked (0.3% hydrogen peroxide in PBS), after which sections were permeabilized with 1% Triton X-100 for 10 min. After rinsing 3 times with PBS for 5 min, the sections were blocked using 2% BSA for 1 h at room temperature and incubated with α-SMA (1:500) at 4°C overnight. After 3 rinses with PBS for 10 min, the sections were further incubated with 3, 3N-Diaminobenzidine tertrahydrochloride (DAB) solution. The reaction product was then developed with diaminobenzidine for 1 min and examined under a light microscope (TH4-200, Olympus, Japan).
Immunofluorescence staining
Cryosections (5 mm thick) were fixed in acetone for 10 min. The samples were washed 3 times with PBS for 10 min per wash and then incubated in 0.2% Triton X-100 for 10 min. After rinsing the sections 3 times in PBS for 5 min, the samples were blocked with 2% BSA for 1 h at room temperature and incubated with primary antibodies CD31 (1:200) overnight at 4°C. The next day, samples were washed 3 times in PBS for 10 min and incubated with Alexa Fluor 594-labeled goat anti-rabbit IgG antibody (1:1,000). After 3 washes in PBS and counterstaining with DAPI (No. 28718-90-3), a laser confocal scanning microscope (TH4-200, Olympus, Japan) was used to study the immunofluorescence.
Statistical analysis
Mean ± SEM data were presented in all instances. Statistical analyses were conducted using GraphPad Prism version 8 software (GraphPad Software Inc., San Diego, CA, USA). A one-way analysis of variance (ANOVA) was utilized for comparing multiple experimental groups, followed by Tukey’s honestly significant difference test for post hoc comparisons to control the family-wise error rate. Alternatively, two-way ANOVA with Šídák’s multiple comparisons test was employed when appropriate. For comparing 2 groups, the Student’s t-test was employed. Any P value below 0.05 was considered statistically significant.
Results
AZD6738 decreases HUVECs viability
First, we investigated the effect of AZD6738 on cell viability. As shown in Fig. 1A, there was no significant difference in cell viability between the control, 0.1% DMSO, and the groups treated with lower concentrations of AZD6738 (0.25 and 0.50 μM) at both 24 and 48 h. As the concentration of AZD6738 increased from 1.00 to 10.00 μM, a gradual and statistically significant decrease in cell viability was observed. Moreover, the cell viability at 48 h was significantly lower than that at 24 h (P < 0.001), indicating a time-dependent effect. The highest concentration (10.00 μM) showed the most pronounced reduction in cell viability, indicating a dose-dependent cytotoxic effect of AZD6738 on HUVECs. And the concentrations of 1 and 5 μM were selected for further investigations.
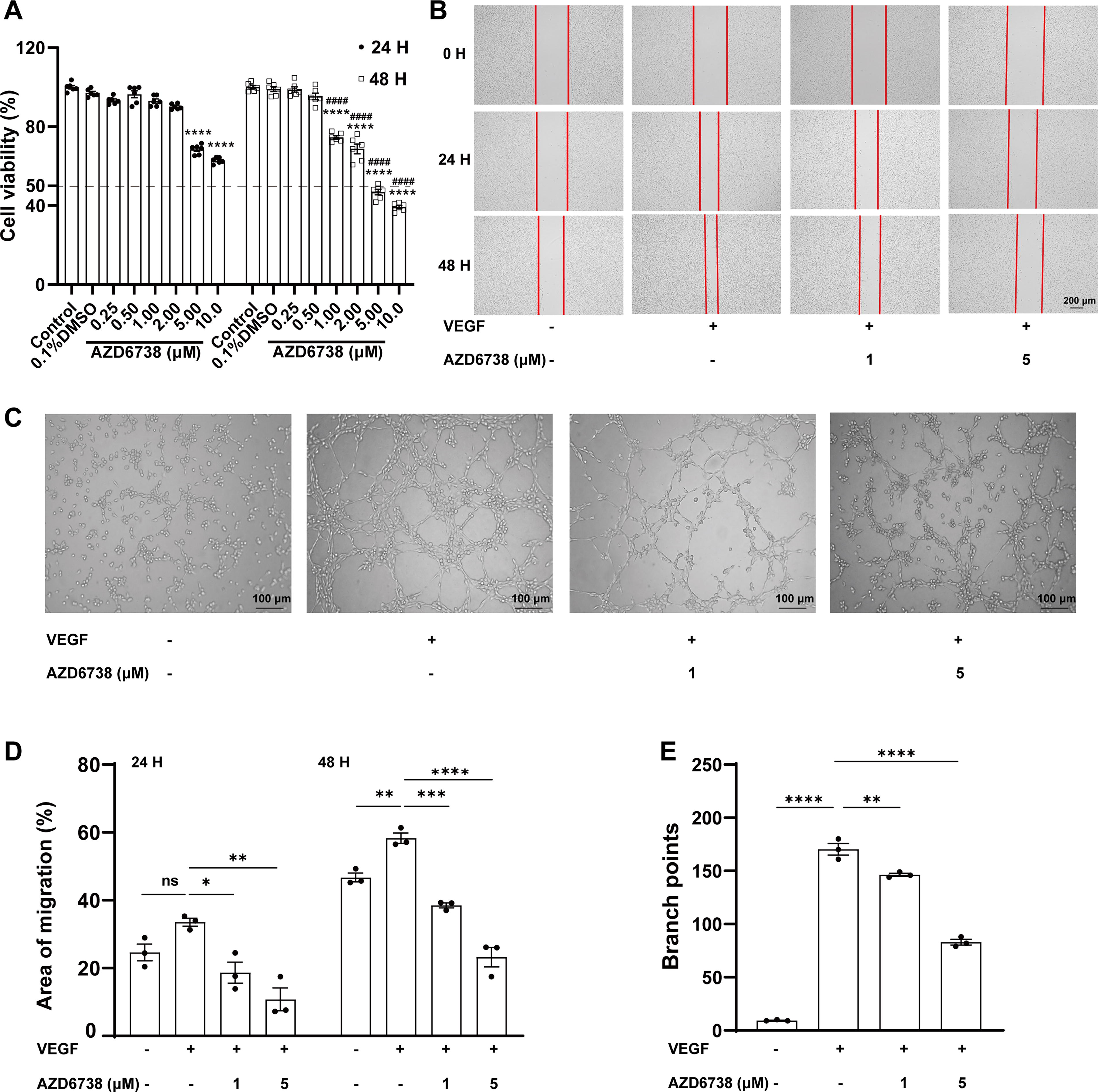
AZD6738 inhibited VEGF-induced HUVECs viability, migration, and capillary structure formation.
AZD6738 inhibited VEGF-induced migration of HUVECs
Then we further assessed the impact of AZD6738 on VEGF-induced cell migration over time. Figure 1B demonstrated that cells treated with VEGF alone exhibited significant migration and wound closure over time. However, when AZD6738 was introduced, there was a clear inhibition of cell migration. Figure 1D quantitatively confirmed these observations. Specifically, in the VEGF group, the migration rates were 33.53% ± 1.18% at 24 h and 58.32% ± 1.53% at 48 h. The addition of AZD6738 at both concentrations resulted in a substantial reduction in the migration area. At 1 μM, the inhibition rates were 44.2% (P < 0.05) at 24 h and 34.0% (P < 0.01) at 48 h. At 5 μM, the inhibition rates further increased to 68.0% (P < 0.001) at 24 h and 60.1% (P < 0.0001) at 48 h. The statistical analysis showed that the addition of AZD6738 at both concentrations resulted in a substantial reduction in the migration area, with the inhibitory effect being more pronounced at 48 h and at the higher concentration (5 μM). Additionally, concurrent treatment with the autophagy inhibitor chloroquine led to a less prominent inhibition of cell migration, suggesting a potential interaction between AZD6738 and the autophagic process (Supplementary Fig. S1A, B).
AZD6738 decreased VEGF-induced tube formation of HUVECs
As for the tube formation ability, the images in Fig. 1C demonstrated that cells treated with VEGF alone formed extensive and complex tubular networks. However, when AZD6738 was added, there was a clear disruption in tube formation, with fewer and less intricate structures observed. Figure 1E quantitatively confirmed these observations. The statistical analysis showed that the addition of AZD6738 at both concentrations led to a marked reduction in branch points, with the inhibitory effect being more pronounced at the higher concentration (5 μM). Additionally, co-treating HUVECs with CQ resulted in an enhancement of tube formation (Supplementary Fig. S1C, D).
AZD6738 decreased VEGF-induced cell proliferation and migration
Cell proliferation was assessed using EdU labeling, and migration was evaluated using transwell migration assays. We investigated the effects of AZD6738 and rapamycin on VEGF-induced cell proliferation and migration in HUVECs. Both AZD6738 and rapamycin significantly inhibited VEGF-induced proliferation and migration of HUVECs. However, rapamycin demonstrated greater efficacy than AZD6738 at a concentration of 20 μM. These findings suggest that while both compounds have antiangiogenic potential, rapamycin may be more effective in suppressing HUVEC cell proliferation and migration under the tested conditions (Supplementary Fig. S2A–C).
AZD6738 enhances autophagy and inhibits the PI3K-AKT signaling pathway in VEGF-elicited HUVECs
Then, we investigated the effect of AZD6738 on autophagy. The immunofluorescence staining of the autophagy marker MDC, as depicted in Fig. 2A, revealed that the VEGF-treated group displayed a prominent increase in MDC-positive staining. Notably, treatment with AZD6738 at concentrations of 1 μM (AZD-1) and 5 μM (AZD-5) led to a further augmentation of MDC staining compared with both the control and VEGF groups (Fig. 2A).
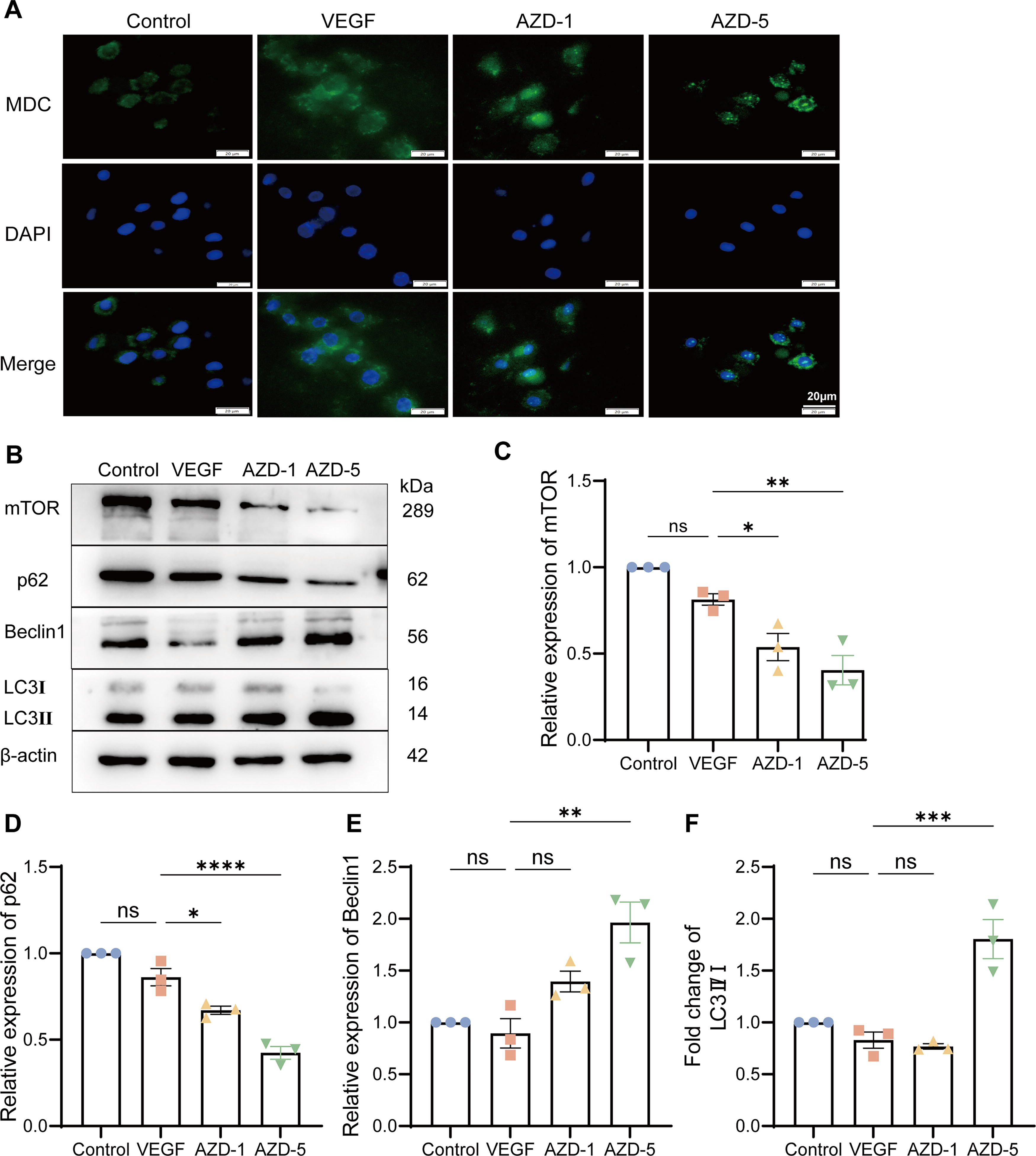
AZD6738 treatment leads to autophagy accumulation.
Subsequently, the levels of autophagy-related proteins were evaluated. As illustrated in Fig. 2B, C, treatment with AZD6738 at 1 and 5 μM resulted in a more pronounced decrease in mTOR expression compared with the VEGF group, indicating that AZD6738 might enhance autophagy by further inhibiting the mTOR signaling pathway. The expression of p62, an autophagy substrate, was slightly reduced in the VEGF group in contrast to the control. However, treatment with AZD6738 at 5 μM led to a more substantial decrease in p62 levels, which may imply a reduction in autophagic degradation (Fig. 2D). Beclin1, a crucial regulator of autophagy initiation, showed an increased expression upon treatment with AZD6738 at 5 μM, suggesting a promotion of autophagy initiation (Fig. 2E). Regarding the LC3 II/I ratio, both the control and VEGF groups exhibited comparable LC3 II/I ratios. Nevertheless, treatment with AZD6738 at 5 μM caused a significant elevation in the LC3 II/I ratio, indicating an enhancement of the autophagic process (Fig. 2F).
Next, we further examined the impact of AZD6738 on VEGFA and VEGFR2 expression as well as the associated signaling pathway. As shown in Fig. 3A, the VEGF-treated group demonstrated elevated expression of VEGF and VEGFR2. Interestingly, treatment with AZD6738 at 5 μM (AZD-5) led to a marked reduction in the expression levels of VEGF and VEGFR2 (Fig. 3B, C), along with a decrease in the phosphorylation of AMPK and AKT, suggesting that AZD6738 effectively inhibits the PI3K-AKT-VEGF signaling pathway (Fig. 3D, E).
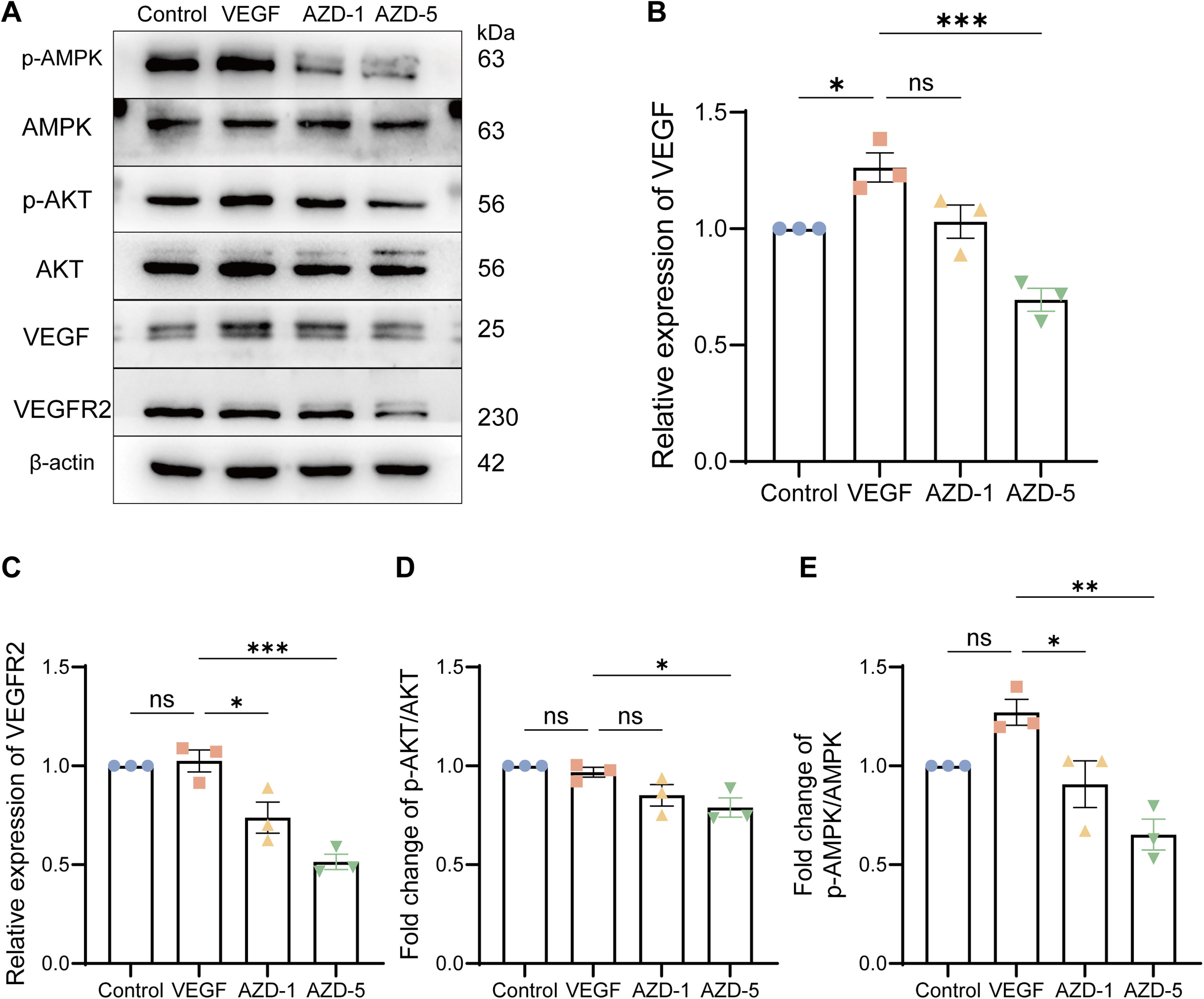
AZD6738 blocks activation of the PI3K-Akt-VEGF pathway.
AZD6738 decreases angiogenesis and opacity in the alkali burn rats by regulating autophagy
AZD6738 decreases angiogenesis and opacity in the alkali burn rats
We visually assessed the effect of AZD6738 on CNV in an experimental model. The control group of normal corneas showed a clear and avascular corneal surface. In the CNV group, significant neovascularization was observed, with blood vessels extending into the cornea. Treatment with AZD6738 noticeably reduced the extent of CNV, with fewer and shorter blood vessels present compared with the CNV group (Fig. 4A, E, F) and Hematoxylin eosin (HE) staining was performed to show structural changes (Fig. 4B, G). The control group showed normal corneal architecture without any signs of neovascularization. In the CNV group, there was clear evidence of neovascularization, with new blood vessels present within the corneal stroma. Treatment with AZD6738 led to a significant reduction in the number and size of these new vessels, as well as less inflammation. Besides, the immunohistochemical (IHC) staining for α-SMA, a marker for myofibroblasts and vascular smooth muscle cells, together with the immunofluorescence (IF) staining for CD31, an endothelial cell marker, collectively indicated that AZD6738 could not only reduce endothelial cell proliferation and neovascularization but also inhibit fibrosis process (Fig. 4C, D).
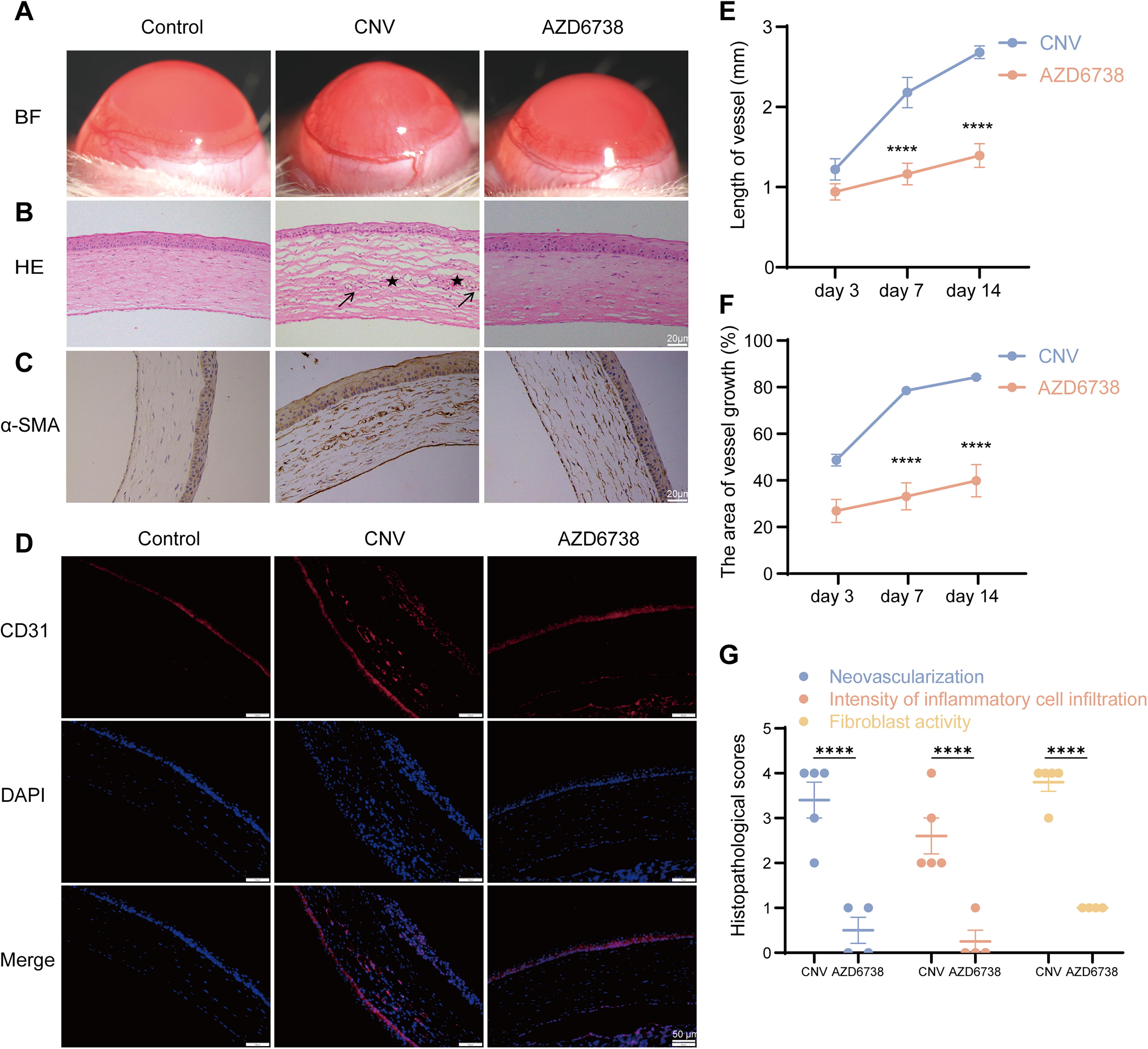
The in vivo antiangiogenic property of AZD6738 in alkali burn-induced corneal neovascularization was investigated in Sprague-Dawley rats.
AZD6738 decreases angiogenesis by regulating autophagy protein, VEGF and VEGFR2
Finally, we focused on the effects of AZD6738 in a CNV animal model as well as involved mechanism. Compared with normal CNV group, treatment with AZD6738 in the CNV model resulted in a significant decrease in mTOR and p62 expression, with relative expression levels reduced by 47.16% and 29.22% (Fig. 5A, C, D; P < 0.05). Conversely, Beclin1 expression and the LC3 II/I ratio increased, indicating enhanced autophagy. The relative expression of Beclin1 increased by 92.9% (Fig. 5A, E; P < 0.05), and the LC3 II/I ratio was elevated by 120.1% compared with the CNV group (Fig. 5A, F; P < 0.0001). At the same time, treatment with AZD6738 resulted in a marked reduction in the expression of VEGFR2 and VEGF, supporting its inhibitory effect on angiogenesis. The relative expression levels of VEGFR2 and VEGF were decreased by 49.82% and 55.29%, respectively (Fig. 5B, H, I; P < 0.05). The mRNA expression levels of VEGF and VEGFR2 were consistent with the protein levels, with the relative mRNA expression of VEGF reduced by 85.00% and that of VEGFR2 by 261.30% in the AZD6738-treated group compared with the CNV group (Fig. 5J, K; P < 0.05). These findings collectively highlight the multifaceted effects of AZD6738 on autophagy and angiogenesis-related pathways. As for involved signaling pathway, analysis of Fig. 5G demonstrated that the p-AKT/AKT ratio was increased in the CNV group, reflecting activation of the PI3K-AKT pathway. However, AZD6738 treatment significantly decreased this ratio by 43.45%, indicating suppression of the pathway (Fig. 5G; P < 0.01), while AZD6738 treatment significantly decreased this ratio, suggesting suppression of the pathway.
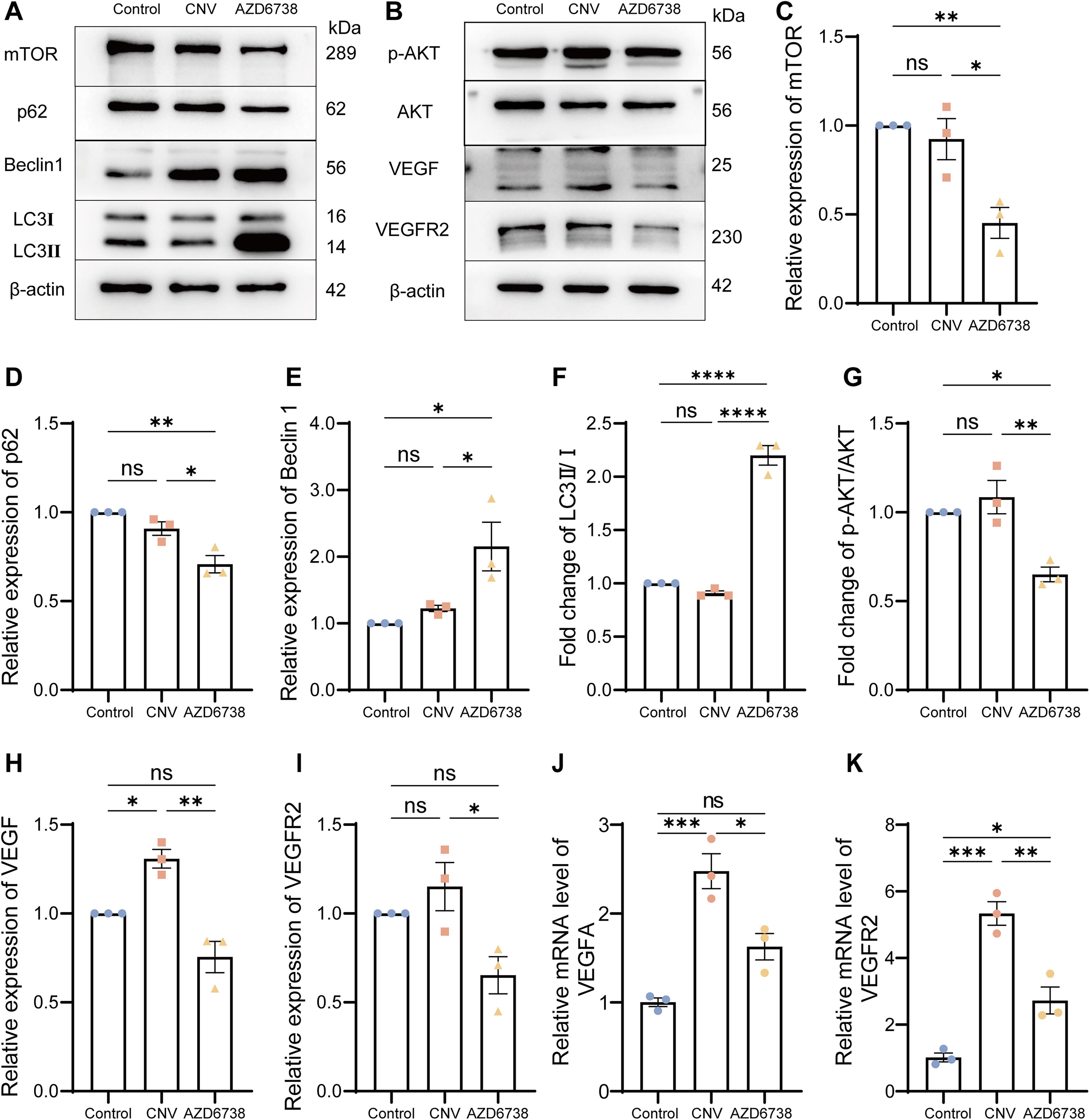
AZD6738 blocks activation of the PI3K/AKT pathway and triggers autophagy.
Discussion
In this study, we demonstrated that AZD6738, an ATR inhibitor, exerts significant antiangiogenic effects in a CNV model by potently enhancing autophagy and simultaneously inhibiting PI3K/AKT signaling pathway. Our results highlight the dual role of AZD6738 in enhancing autophagic activity while simultaneously inhibiting the PI3K/AKT pathway, contributing to its efficacy in reducing CNV.
Autophagy plays a crucial role in maintaining cellular homeostasis, particularly in the context of inflammation and immune responses. 29 The activation of autophagy has been shown to suppress pathological angiogenesis, as seen in previous studies where enhanced autophagy mitigated CNV and prolonged corneal graft survival by inhibiting inflammation.30,31 Our findings are consistent with these observations, as AZD6738 treatment led to increased expression of LC3-II and Beclin1, along with a significant reduction in p62 levels, both in vitro and in vivo. These changes in autophagic markers indicate that AZD6738 promotes autophagic flux, which may contribute to its antiangiogenic effects. Moreover, the suppression of mTOR signaling by AZD6738 further supports the activation of autophagy, aligning with the well-established role of mTOR as a negative regulator of autophagy.21,32–34
In addition to its effects on autophagy, AZD6738 significantly inhibited the PI3K/AKT pathway, a critical signaling cascade involved in cell proliferation, survival, and angiogenesis. 35 Our data demonstrated that AZD6738 reduced the phosphorylation of AKT, leading to decreased VEGF and VEGFR2 expression, which are key mediators of angiogenesis. This finding is particularly relevant given that the PI3K/AKT pathway is often upregulated in various angiogenic processes, including CNV.17,18,21,36 By inhibiting this pathway, AZD6738 effectively disrupts the pro-angiogenic signaling that underlies CNV pathogenesis, thereby reducing neovascularization.
Notably, a recent study demonstrated that AZD6738 attenuates lipopolysaccharide (LPS)-induced corneal inflammation and fibrosis by modulating macrophage function and polarization. 37 While this study utilized a different corneal pathology model, both models involve dysregulated corneal homeostasis, highlighting the broad therapeutic potential of AZD6738 in corneal diseases. The drug’s efficacy in reducing inflammation, a process tightly linked to neovascularization in ocular disorders, suggests that its anti-inflammatory actions may complement its direct effects on autophagy and PI3K/AKT signaling to inhibit CNV. Inflammatory cells and cytokines, such as those regulated by AZD6738 in the LPS model, can promote vascular endothelial cell activation and neovascular sprouting, meaning that dampening inflammation could further disrupt the pro-angiogenic microenvironment in CNV. 38
Autophagy degrades components of the nod-like receptor protein 3 (NLRP3) inflammasome, thereby inhibiting its activation and reducing the release of pro-inflammatory cytokines such as interleukin-1β (IL-1β). 39 Additionally, autophagy modulates the activity of transcription factors like nuclear factor kappa-B (NF-κB), influencing the expression of inflammation-related genes. 40 The activation of autophagy suppresses the polarization of M1 macrophages. 41 By clearing the NLRP3 inflammasome, autophagy prevents its hyperactivation, dampens the inflammatory response, and promotes the polarization of M2 macrophages, highlighting autophagy’s crucial role in balancing inflammation and macrophage function. 42 This intricate interplay highlights the crucial role of autophagy in maintaining a delicate balance between inflammation and macrophage function, which is likely integral to the therapeutic effects of AZD6738 in corneal pathologies.
The clinical implications of our findings are significant, particularly in the context of developing new therapeutic strategies for CNV, which remains a challenging condition with limited treatment options. 43 The ability of AZD6738 to simultaneously target autophagy and the PI3K/AKT pathway positions it as a highly promising candidate for CNV therapy. 25 Moreover, our study builds on existing research by providing a novel insight into the use of ATR inhibitors in ocular diseases, a potential application that has not been extensively explored.
Despite the promising results, our study has several limitations that should be acknowledged. First, while we demonstrated the effects of AZD6738 on autophagy and the PI3K/AKT pathway in vitro and in a CNV model, the exact molecular mechanisms through which AZD6738 modulates these pathways require further investigation. Second, our study focused primarily on short-term outcomes; therefore, the long-term efficacy and safety of AZD6738 in treating CNV remain to be determined. Additionally, the broader applicability of AZD6738 across different models of ocular neovascularization needs to be explored to fully understand its therapeutic potential. Future studies should aim to address these limitations and validate our findings in clinical settings.
Conclusion
Our study demonstrates that AZD6738, an ATR inhibitor, holds significant promise as a therapeutic agent for the treatment of CNV. The efficacy of AZD6738 has been validated in both in vitro and animal models, highlighting its potential for clinical application. However, a deeper understanding of the molecular mechanisms underpinning the antiangiogenic effects of ATR inhibition is essential, underscoring the need for further research in this area.
Footnotes
Acknowledgments
The authors would like to express their gratitude to all the members of the Jiangxi Key Laboratory of Clinical Pharmacokinetics for their valuable contributions through daily discussions and advice.
Authors’ Contributions
Q.W. performed design, investigation, data acquisition, validation, and writing original draft of the article. Y.P., together with J.X. and C.C., was responsible for formal analysis of the data and reviewing the article. X.L., together with K.Y., W.Y., Y.P., and Z.Z., was responsible for the validation, software, conceptualization, and supervision. Y.Y. and S.W. were responsible for funding acquisition, project administration, reviewing, and approving the final version of the article. All the authors declare that they have no conflicts of interest with the contents of this article.
Availability of Data and Materials
The data generated and analyzed during this study are available from the corresponding author upon request.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the Natural Science Foundation of Jiangxi Province (No. 20212ACB2060022), the Chinese Medicine Science and Technology Project of Jiangxi Province (2019A1889), the Science and Technology Project of Jiangxi Provincial Health Commission (202110043), the Yingtan Science and Technology Program (20244-380332), and the Graduate Innovation Special Fund (YC2023-B099).