
Abstract
Abstract
Pyloric atresia (PA) is rare and may be associated with epidermolysis bullosa (EB). This is the first case report of a successful laparoscopic treatment of PA in a full-term 7-day-old neonate with EB. The laparoscopic approach consists of a longitudinal pyloromyotomy and excision of the thick obstructing pyloric membrane with a Heineke–Mickulicz pyloroplasty closure. Oral feeding was resumed at postoperative Day 7, and the child was discharged 5 days later with satisfactory follow-up at 8 months. Recommendations are provided for the management of the neonate with PA/EB.
Introduction
Case Report
A full-term newborn girl with a prenatal sonogram of polyhydramnios was delivered by cesarean section at a provincial hospital. The patient passed normal meconium in the first day after birth. She subsequently had nonbilious emesis and respiratory distress during feeding. She also developed multiple skin erosions, especially at her wrists and ankles, and was referred to our center on the second day of life for further management. The family history is significant for a sister with PA and EB, who underwent open duodenogastrostomy as a newborn in our center 13 months ago.
The diagnostic work-up consisted of plain abdominal X-ray showing a large gastric bubble with no air in the intestines and an upper gastrointestinal contrast study demonstrating a markedly dilated stomach with complete pyloric obstruction (Fig. 1), confirming PA. Erosions and bullous epidermal lesions consistent with EB continued to evolve on her limbs, face, and abdomen. The patient was admitted to the intensive care unit for the management of her respiratory distress, indirect hyperbilirubinemia, and EB wound care.
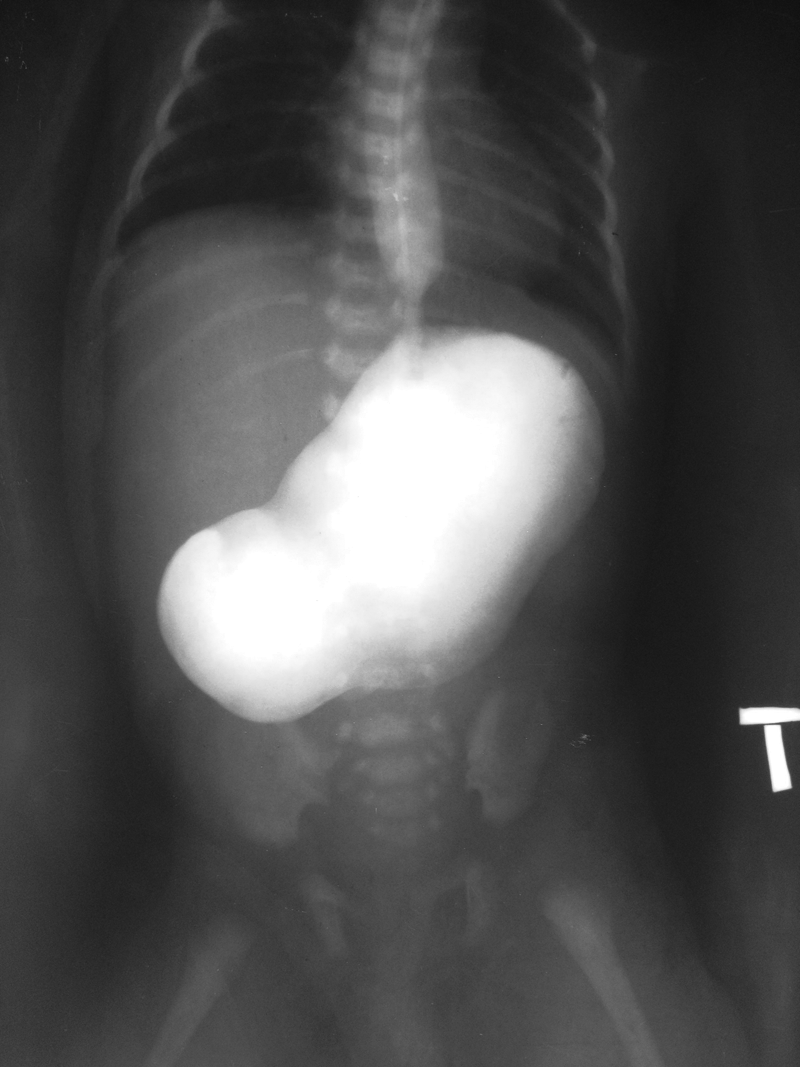
Upper gastrointestinal study showed complete pyloric obstruction with gastric dilatation.
Soon after stabilization of her general condition, the child was operated on at Day 8 of age. Her body weight at the time of surgery was 2000 g. The laparoscopic procedure was performed using one infraumbilical 5-mm port for the camera and two operating 3-mm ports at the right and left lower abdomen. CO2 insuflation was maintained at a pressure of 8 mm Hg and a flow of 2.5 L/minute. The stomach was markedly dilated, and the pylorus and duodenum are contracted. A 1.5-cm longitudinal incision was made on the pylorus, and the thick 4–5-mm pyloric membrane causing complete pyloric obstruction was excised. A Heineke–Mickulicz pyloroplasty was performed with a one-layer interrupted intracorporeal closure using 5.0 polydioxanone suture. The blood loss was minimal. The operative time was 90 minutes.
The patient recovered well after the operation. Oral feeding was started at postoperative Day 7, and she was discharged 5 days later. At 8 months of follow-up, the child was doing well, tolerating enteral feed, gaining weight, and receiving careful care for her skin lesions.
Discussion
The most common differential diagnosis of PA is duodenal atresia and, more rarely, gastric antral web. Although PA patients always present with nonbilious emesis, 1 most duodenal atresia patients have bilious emesis. The clinical manifestation can be, however, confounding in cases of duodenal atresia where the duodenal obstruction is proximal to Vater's ampulla. Plain abdominal X-ray can help differentiate between the two entities showing the typical single gastric bubble for PA 10 and the double bubble for duodenal atresia. An upper gastrointestinal contrast study showing arrest of contrast passage from the stomach is helpful in determining the diagnosis. Gastric antral web often presents in older children, and barium upper gastrointestinal series demonstrate classic features of double bulb appearance: the normal duodenal bulb and a proximal antral chamber between the web and the pylorus. 11
A neonate presenting with nonbilious vomiting and multiple skin erosions or bullous lesions should be suspected of having PA associated with EB. The two forms of EB associated with PA are EB simplex and junctional EB. 2 The severity of skin involvement in EB/PA can be highly variable. 2 Although mutations of 10 different genes underlie the pathology of EB, EB associated with PA has been reported to be caused by mutation of the plectin gene (PLEC1) or intergrin genes (ITGA6, ITGB4) 2 with an autosomal recessive inheritance. 2 Genetic investigations were not performed for this patient and her sibling to determine the mode of inheritance.
PAs are classified into three types: type 1, associated with a membrane; type 2, with solid fibrous pyloric cord; and type 3, with a complete gap between stomach and duodenum. PA type 1 is treated by surgical excision of the membrane and a Heineke–Mickulicz pyloroplasty, whereas PA types 2 and 3 are repaired with gastroduodenostomy bypass.1,6 The results are usually good with the surgical treatment for simple PA. 6 PA-EB prognosis is, however, poor with lethal outcome in the majority of cases because of the complications of malnutrition and/or infection.6–8 This is the first report of successful management of PA in general and, in particular, of PA-EB using laparoscopic repair. We postulate that early restoration of intestinal continuity and the use of a minimally invasive approach for PA in an EB patient who also has massive metabolic needs from her EB wounds may have an advantage over the open laparotomy. The laparoscopic approach may facilitate earlier enteral nutrition and reduce the EB-associated risks for wound infection and wound repair morbidities of the open approach. Although the experience is limited to 1 case, our result suggests that laparoscopic surgery could be safe and effective in the treatment of PA in infants with EB.
Footnotes
Acknowledgments
We would like to thank Professor Ai-Xuan Le Holterman and the IPSAC-VN for their valuable comments and editing of this manuscript.
Disclosure Statement
No competing financial interests exist.