
Abstract
Abstract
Background:
Inflammatory cytokines dysregulate microvascular function, yet how cytokines affect lymphatic endothelial cells (LEC) are unclear.
Methods and Results:
We examined effects of TNF-α, IL-1β, and IFN-γ on LEC proliferation, endothelial cell adhesion molecule (ECAM) expression, capillary formation, and barrier changes in murine (SV-LEC) and human LECs (HMEC-1a).
Results:
All cytokines induced ICAM-1, VCAM-1, MAdCAM-1, and E-selectin in SV-LECs; TNF-α, IL-1β and IFN-γ induced ECAMs (but not MAdCAM-1) in HMEC-1a. IL-1β increased, while IFN-γ and TNF-α reduced SV-LEC proliferation. While TNF-α induced, IFN-γ decreased, and IL-1β did not show any effect on HMEC-1a proliferation. TNF-α, IL-1β, and IFN-γ each reduced capillary formation in SV-LEC and in HMEC-1a. TNF-α and IL-1β reduced barrier in SV-LEC and HMEC-1a; IFN-γ did not affect SV-LEC barrier, but enhanced HMEC-1a barrier. Inflammatory cytokines alter LEC growth, activation and barrier function in vitro and may disturb lymphatic clearance increasing tissue edema in vivo.
Conclusion:
Therapies that maintain or restore lymphatic function (including cytokines blockade), may represent important strategies for limiting inflammation.
Introduction
Lymphatic endothelial cells (LEC) are functionally, biochemically, and developmentally distinct from blood endothelial cells (BEC). 6 In ‘lymphangiogenesis,’ new lymphatic capillaries can be generated by sprouting, migration, and proliferation from pre-existing LEC or recruitment of LEC progenitors.7–10 Changes in lymphatic function and proliferation alter interstitial fluid absorption and homeostasis, leukocyte and tumor cell traffic in tumor metastasis, lymphedema, and inflammation.11–13 After development, lymphangiogenesis14,15 is active during tissue repair, regeneration, and inflammation. 16 Although BEC and LEC may respond to equivalent physiological levels of cytokines and exhibit similar responses in survival, migration, and proliferation, unregulated or supraphysiological increases in cytokines may initiate or drive inflammation-associated pathological events. Lymphangiogenesis17,18 might however have different effects on inflammation as compared to angiogenesis. For example, ECAMs expression on inflamed venules facilitate leukocyte extravasation into tissues.19,20 ECAM induction on 1° lymphatics might support the transfer of leukocytes and antigen-presentation cells to lymph nodes. 21
Mediators that induce and regulate lymphatics are not yet defined, but an improved understanding of the role(s) played by modulation of lymphatic endothelial integrity could reduce inflammatory edema produced by LEC dysfunction. For example, TNF-α can induce BEC inflammation and edema, and TNF-α suppression enhances lymphangiogenesis. 22 Although cytokines such as TNF-α participate in inflammatory angiogenesis in several clinical conditions,23–25 cytokines may also regulate lymphatic responses, and play roles in different phases of inflammation that are not yet well understood. To overcome limitations encountered with in vivo lymphangiogenesis models, we used human and mouse LECs to study lymphatic responses to inflammatory cytokines. We found that Th1 cytokines modulate ECAM expression, proliferation, capillary formation and barrier in mouse SV-LECs and human HMEC-1a. LEC, like BEC, are dysregulated by inflammatory cytokines and suggest that cytokine-mediated LEC dysfunction exacerbates inflammation, especially in disorders such as Crohns' disease and ulcerative colitis.
Materials and Methods
Cell culture
Our described mouse (SV-LEC) and human (HMEC-1a) cell lines were cultured as reported previously.26,27
Proliferation
TNF-α, IL-1β, and IFN-γ were purchased from Thermo/Fisher (Waltham, MA). 10% confluent cultures were treated with test agents in medium assaying at time points up to 72 h. LEC growth in response to cytokines was assessed by MTT assay. 28 Cells without cytokines were used as 100% ‘control’ level of cell proliferation with N = 8 replicates.
LEC capillary tube formation assay
Capillary formation on Matrigel (BD Biosciences, San Jose, CA)was used to examine LEC responses to cytokines. 29 Confluent LEC were incubated with cytokines in 10% DMEM for 72 h, loaded with calcein 2-AM (2 μg/ml, 30’) trypsinized, and plated onto Matrigel for 4 h. Capillaries were quantitated as described 29 by counting the number of lymphatic capillaries from central focal planar digital micrographs.
ECAM expression in LEC cells
ECAM expression was assessed as described. 26 LEC were treated with TNF-α, IL-1β, or IFN-γ for 24 h, and incubated with respective anti-ECAM antibody, followed by HRP-tagged 2° antibody. ECAM expression was analyzed and expressed as the % maximum (TNF-α induced) level of absorbance at 450 nm. All protocols were performed at least n = 4 replicates.
LEC trans-endothelial electrical resistance (TEER) in response to cytokines
LEC barrier was measured by TEER. LECs were cultured on 8 μm transwells and electrical resistance changes developed LEC monolayers in response to cytokines were measured 0–72 h using an Epithelial Volt Ohm Meter (WPI, Sevenage, United Kingdom).
Statistics
Data were analyzed for statistical differences using Instat (Graphpad Software, La Jolla, CA) for one-way ANOVA with Bonferroni post-testing.
Results
Effects of cytokines on lymphatic endothelial cell proliferation
SV-LEC
At 72 h, TNF-α and IFN-γ dose-dependently inhibited, while IL-1β dose-dependently increased proliferation. At 72 h, TNF-α decreased proliferation to 70 ± 2% of control at 5 ng/ml (***p < 0.001), to 57 ± 2% of control at 10 ng/ml (***p < 0.001), and to 44 ± 2% of control at 20 ng/ml (***p < 0.001) concentrations. We found all concentrations of IFN-γ (100, 500, and 1000 U/ml) decreased LEC proliferation. IFN-γ decreased proliferation to 76 ± 2% of control at 100 U/ml (***p < 0.001), to 73 ± 2% of control at 500 U/ml (***p < 0.001), and to 71 ± 2% of control at 1000 U/ml (***p < 0.001). IL-1β increased proliferation 18% over control at 5 ng/ml (118 ± 3% of control, *p < 0.05), 16% over control at 10 ng/ml (116 ± 4% of control, *p < 0.05), and 14% over control at 20 ng/ml (114 ± 3%, *p < 0.05) (Fig. 1).

SV-LEC proliferation and cytokines. (
HMEC-1a
At 72 h, IFN-γ dose dependently decreased lymphatic proliferation whereas IL-1β did not alter LEC proliferation. Interestingly, TNF-α significantly increased cell proliferation at low concentration; this effect was not seen at higher concentrations. At 72 h, TNF-α increased proliferation to 123 ± 6.80% of control at 5 ng/ml (**p < 0.01), to 116 ± 5.30% (ns) at 10 ng/ml, and to 101.74 ± 6.81% (ns) at 20 ng/ml concentrations. IL-1β had no effect on human LEC proliferation. At 72 h, IL-1β did not affect proliferation (105.45 ± 4.41% of control at 5 ng/ml (ns), 94.75 ± 3.24% of control at 10 ng/ml, and 90.87 ± 6.45% at 10 ng/ml). IFN-γ significantly decreased LEC proliferation at all concentrations. At 72 h, IFN-γ decreased proliferation to 69.04 ± 3.13% of control at 100 U/ml (**p < 0.01), to 61.78 ± 5.19% to that of control at 500 U/ml (**p < 0.01) and almost to half, (50.9 ± 7.63% at 1000 U/ml **p < 0.01) (Fig. 1).
Cytokine-induced LEC adhesion molecule expression
Previous studies show that cytokines induce LEC ICAM-1 and VCAM-1 in LEC in vivo 21 at concentrations and kinetics reported to mobilize ECAMs in BEC.3,26 We therefore determined ECAM expression in LEC.
SV-LEC
ICAM-1 surface expression
TNF-α (20 ng/ml) induced ICAM-1 expression (i.e.,100 ± 6% ‘maximum’ ICAM-1 expression, ***p < 0.001); IL-1β (10 ng/ml) induced ICAM-1 expression to 86 ± 1% of expression, (***p < 0.001); IFN-γ (1000 U/ml) induced ICAM-1 expression to 138 ± 7% maximum of TNF-α induced levels of ICAM-1 expression, (***p < 0.001) (Fig. 2a).
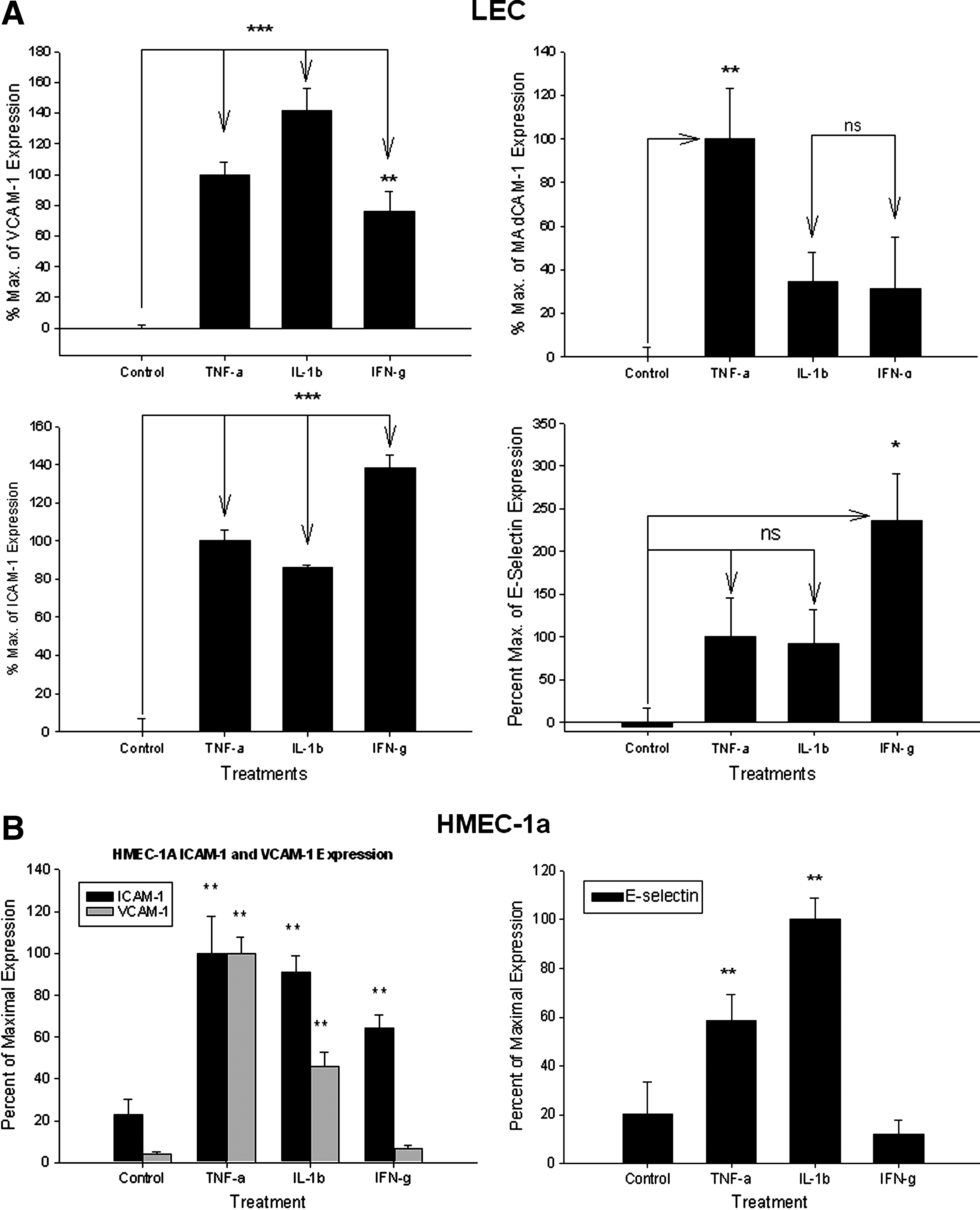
VCAM-1 surface expression
TNF-α (20 ng/ml) induced VCAM-1 expression to 100 ± 8% maximum, (***p < 0.001); IL-1β (10 ng/ml) significantly induced VCAM-1 expression (141 ± 15% maximum, ***p < 0.001); IFN-γ (1000 U/ml) induced VCAM-1 expression (75 ± 14% maximum, **p < 0.01) (Fig. 2a).
MAdCAM-1 surface expression
TNF-α (20 ng/ml) induced MAdCAM-1 expression [set as 100 ± 23% (i.e., ‘maximum’), **p < 0.01]; IL-1β (10 ng/ml) did not induce MAdCAM-1 (34 ± 13% maximum, n.s.; IFN-γ (1000 U/ml) did not induce MAdCAM-1 expression (31 ± 24% maximum, ns) (Fig. 2a).
E-Selectin surface expression
Neither TNF-α (20 ng/ml) (100 ± 46% maximum) or IL-1β (10 ng/ml) significantly affected E-selectin expression (92 ± 40% maximum). Only IFN-γ (1000 U/ml) induced E-selectin expression on LEC (to 235 ± 55% maximum, *p < 0.05) (Fig. 2a).
HMEC-1a
ICAM-1 expression
TNF-α (20 ng/ml) induced ICAM-1 (100 ± 17.7% maximum, **p < 0.01). IL-1β (10 ng/ml) also induced ICAM-1 (90.68 ± 7.92% maximum, **p < 0.01); IFN-γ (1000 U/ml) induced ICAM-1 (but to only 64.34 ± 6.16% maximum, **p < 0.01) (Fig. 2b).
VCAM-1 expression
TNF-α (20 ng/ml) induced VCAM-1 expression (100 ± 7.94% maximum, **p < 0.01); IL-1β (10 ng/ml) also induced VCAM-1 (46.17 ± 6.77% maximum, **p < 0.01); IFN-γ (1000 U/ml) did not alter expression of VCAM-1 on HMEC-1a, and was different from its effect seen on SV-LEC (6.79 ± 1.64% maximum of VCAM-1) (Fig. 2b).
E-selectin expression
IL-1β showed the greatest induction of E-selectin on HMEC-1a cells. TNF-α (20 ng/ml) significantly induced E-Sel expression (58.75 ± 10.32% maximum, **p < 0.01); IL-1β (10 ng/ml) significantly induced E-Sel (100 ± 8.77% maximum, **p < 0.01); IFN-γ (1000 U/ml) did not significantly affect the expression of E-Selectin on HMEC-1a cells, also different from its effect seen on SV-LEC (only 11.91 ± 5.93% maximum, p > 0.05) (Fig. 2b).
MAdCAM-1 expression
No expression of surface MAdCAM-1 expression was observed in response to any of these cytokines on HMEC-1a (data not shown). Neither VEGF-C nor D (100 ng/ml, 24 h) increased ECAMs expression on human or mouse LEC (data not shown)
Effect of cytokines on LEC capillary formation. Cytokines reduce SV-LEC capillary formation
TNF-α and IL-1β significantly reduced LEC capillaries only at higher concentrations, while IFN-γ reduced capillary formation at all concentrations. TNF-α decreased LEC lymphatic capillaries by 19% at 5 ng/ml to 81 ± 12% of control (100%), (not sig.), by 30% 10 ng/ml (to 70 ± 4% of control (100%), n.s.), and 44% at 20 ng/ml to 56 ± 6% of control (100%, **p < 0.01). IL-1β decreased the number of SV-LEC LEC capillaries by 19% (5 ng/ml) to 81 ± 9% of control, (n.s.), 34% at 10 ng/ml to 66 ± 10% of control (100%), (n.s.), and 51% at 20 ng/ml (to 49 ± 11% of control, **p < 0.01). IFN-γ decreased the number of lymphatic capillaries by 87% at 100 U/ml (to 13 ± 3% of control, ***p < 0.001), 85% at 500 U/ml (to 15 ± 3% of control (100%), ***p < 0.001), and 61% at 1000 U/ml (to 39 ± 12% of control, ***p < 0.001) (Figs. 3a and 3b).

Cytokines reduce HMEC-1a lymphatic capillary tube formation
HMEC-1a were analyzed by fluorescent activated cell sorting to check D2-40 expression. 78.6% of HMEC-1a were D2-40+. D2-40+ cells from HMEC-1a were sorted to obtain ∼98.1% pure D2-40+ cells. The D2-40+ cells obtained were used for performing capillary tube formation assay on Matrigel. All concentrations of TNF-α, IL-1β, and IFN-γ tested decreased LEC capillaries. However IFN- γ at higher concentration increased LEC capillary numbers compared to TNF-α and IL-1β (still less than controls). The rank order of lymphatic capillary inhibition was found to be TNF-α ∼ = IL-1β < IFN-γ. TNF-α completely inhibited HMEC-1a capillary formation (at 5 ng, 10 ng, and 20 ng/ml) at 72 h. IL-1β showed the same dose-effect as TNF-α. However IFN- γ at 100 U/ml decreased LEC capillary formation by 16.13 ± 9.31% (**p < 0.01), at 500 U/ml to 8.91 ± 5.16% (**p < 0.01) and at 1000 U/ml to 52.97 ± 2.58 % (**p < 0.01) vs. controls (Figs. 3c and 3d).
Trans endothelial electrical resistance (TEER)
SV-LEC
Control
Control TEER of SV-LEC remained stable without changes in resistance throughout the study (0 h = 100%, 24 h = 103.5 ± 3.51%, 48 h = 103.9 ± 2.23%, 72 h = 94.22 ± 3.97%).
TNF-α
20 ng/ml time-dependently decreased barrier in SV-LEC monolayers (0 h = 100%, 24 h = 91.3 ± 7.5% **p < 0.01, 48 h = 87.8 ± 4.6% **p < 0.01, 72h = 84 ± 2.48 **p < 0.01) (Fig. 4).

SV-LEC and HMEC-1a barrier. SV-LEC TEER of control cells was constant throughout the study. TNF-α and IL-1β time-dependently decreased SV-LEC TEER. IFN-γ did not alter TEER. HMEC-1a TEER. Control TEER of control cells decreased slightly over 72 h. TNF-α: TNF-α decreased TEER at 48–72 h. IL-1β: No significant alterations in HMEC-1a TEER with IL-1b (10 ng/ml). IFN-γ: IFN-γ significantly increased HMEC-1a TEER at 24–72 h.
IL-1β
IL-1β 10 ng/ml time-dependently decreased barrier in SV-LEC monolayers (0 h = 100%, 24 h = 90.1 ± 0.8 **p < 0.01, 48 h = 88.9 ± 4% **p < 0.01, 72 h = 88.5 ± 11.8 **p < 0.01) (Fig. 4).
IFN-γ
IFN-γ did not alter SV-LEC barrier compared with control over 72 h (0 h = 100%, 24 h = 102.5 ± 2.73%, 48 h = 105 ± 2.73% and 72 h = 91 ± 6.3%) (Fig. 4).
HMEC-1a
Control
Untreated HMEC-1a were used as controls. Barrier in control cells decreased significantly from 24 h to 72 h (0 h = 100%, 24 h = 85.95 ± 0.55 **p < 0.01, 48 h = 85.1 ± 1.72 **p < 0.01, 72 h = 91.03 ± 1.26 **p < 0.01) (Fig. 4).
TNF-α
TNF-α at 20 ng/ml did not alter the HMEC-1a barrier at 24 h. Barrier decreased at 48–72 h (0 h = 100%, 24 h = 93.45 ± 3.11, 48 h = 83.6 ± 1.15% **p < 0.01, 72 h = 91.74 ± 1.28% *p < 0.05) (Fig. 4).
IL-1β
No change in HMEC-1a barrier was observed with IL-1β
IFN-γ
IFN-γ increased HMEC-1a barrier at all time points vs. controls or other treatments (0 h = 100%, 24 h = 119.07 ± 1.73% **p < 0.01, 48 h = 108.93 ± 2.25% *p < 0.05, and 72 h = 108.07 ± 1.04% *p < 0.05) (Fig. 4).
Discussion
The diverse physiological functions of BEC and LEC are regulated by several classes of vasoactive mediators. Lymphangiogenesis is regulated by signaling mechanisms, including VEGFs, 6 cytokines,30,31 and environmental cues. 32 Although studies demonstrate expansion of lymphatics within inflamed tissues, 17 whether lymphangiogenesis during inflammation is beneficial or exacerbating is controversial. Besides lymphatic vessel density, characteristics of lymphatic vessels may be affected by amplification of cytokines at inflammatory sites. So far, few studies have considered how cytokine effects on LEC proliferation, capillary tube formation, and ECAM expression might influence inflammation.
Angiogenesis and lymphangiogenesis are complex processes modulated by immune cells, complex networks of cytokines, and VEGFs. Franchi et al. report inflammatory cytokines play a pivotal role in angiogenesis, proliferation, and ECAM expression.33,34 In BEC, TNF-α and IL-1β are angiogenic and upregulate ICAM-1 and VCAM-1. TNF-α has been reported to decrease while IL-1β increases proliferation. IFN-γ inhibits proliferation but upregulates ICAM-1 and VCAM-1.35,36 VEGF-A is itself an inflammatory cytokine that induces ICAM-1 in BEC. 37 VEGF-A supports endothelial survival, proliferation, migration, capillary genesis and permeability, and inflammatory cell recruitment to BEC; VEGF-A also modulates LEC proliferation. 38 VEGF-C released by platelets, monocytes, macrophages, and BEC, 39 also regulates BEC proliferation, vascular permeability, 40 capillary formation, 41 inflammatory-activation, 42 and leukocyte binding to BEC. 37 VEGF-C and VEGF-D also play dominant roles in BEC and LEC survival, proliferation, migration, capillary formation in vivo and in vitro; 43 their effects in inflammation are less well characterized.
Cytokines modulate lymphatic endothelial cell proliferation
VEGFs modulate BEC and LEC proliferation during development and disease. 44 While TNF-α, IL-1β, and IFN-γ modulate BEC proliferation, capillary formation, and ECAM expression, effects of cytokines on LEC are not fully understood. We found that TNF-α, IL-1β, and IFN-γ dose-dependently influence mouse and human LEC. Previously TNF-α has been reported to reduce LEC proliferation.22,29 Interestingly TNF-α inhibited proliferation only at low (5 ng/ml) concentrations in human LEC; higher TNF-α levels did not inhibit growth. IL-1β released by leukocytes, mast cells, and BEC 2 positively regulates BEC survival, proliferation, and angiogenesis in vivo. We found a similar effect in mouse LEC where proliferation was increased by IL-1β at 5–20 ng/ml. Human LEC did proliferate in response to IL-1β. IFN-α/IFN-γ inhibit mouse and human LEC proliferation and might be ‘anti-lymphangiogenic’. 25 Consistent with those findings, we observed all concentrations of IFN-γ decreased human and mouse LEC proliferation.
Cytokine-mediated ECAM surface expression in SV-LEC and HMEC-1a cells
Inflammatory cytokines are central mediators of inflammation and potently induce adhesion molecules on BEC and LEC in vitro.25,45–49 While BEC upregulate ICAM-1, VCAM-1, MAdCAM-1, and E-selectin in response to inflammatory cytokines and VEGF-A, 37 less information is available about the expression of these adhesion molecules on stimulated LEC. In human umbilical vein endothelial cells, VEGF-A and TNF-α synergistically enhance E-selectin expression, which recruits neutrophils to activated endothelium at sites of inflammation. 50 While blood vascular ECAM expression brings leukocytes to sites of inflammation, lymphatic vascular ECAM expression may retain leukocyte populations at sites of inflammation or facilitate entry into lymphatics, potentially maintaining/resolving inflammation by regulating the exit of these leukocytes from inflamed tissues. An improved understanding of the cooperation between adhesive systems on both sides of the vascular system may enhance our ability to coordinate and regulate the entry and exit of leukocyte to control inflammation.
Johnson et al. reported that inflammatory cytokines upregulate LEC ICAM-1, VCAM-1, and E-selectin, and the chemokines MCP-1, RANTES, and MIP-3α.21,36 That study found high ICAM-1 and VCAM-1 induction in response to TNF-α (followed by IL-1β and IFN-γ. Similar studies using human neonatal LEC also found TNF-α dose-dependently increased VCAM-1 and ICAM-1. 51 Furthermore TNF-α induces VCAM-1 and ICAM-1 expression on both BEC and LEC, but at a quantitatively lower level in LEC. Sawa et al. reported that lymphocyte transmigration across lymphatics may not depend on cytokines 52 and that cytokines may influence other LEC behaviors independent of lymphocyte trafficking. Other studies have shown that TNF-α induces expression of ICAM-1, VCAM-1, MAdCAM-1, and E-selectin in the SVEC4-10 endothelial cell line isolated from murine (C3H/Hej) axillary lymph nodes, which represent high endothelial venular endothelial cells.53–55
We compared differences between ECAMs induction by cytokines in mouse and human LEC. In mouse LEC, ICAM-1, VCAM-1 and E-selectin were all potently induced by TNF-α, IL-1β, and IFN-γ. These responses did not show the rank order potency reported by Johnson et al. 21 and may reflect differences in the anatomic origin of these cells (skin vs. mesentery), species or in vitro culture conditions. Only TNF-α (not IL-1β nor IFN-γ) induced MAdCAM-1 in mouse LEC. Human LEC showed different ECAM induction with cytokines. TNF-α and IL-1β increased ICAM-1, VCAM-1, and E-Sel in mouse LEC. IFN-γ induced ICAM-1, but neither VCAM-1 nor E-Sel. Moreover we could not induce MAdCAM-1 with any of cytokines (TNF-α, IL-1β, and IFN-γ) on human LEC. MAdCAM-1 is strongly expressed by high endothelial venules, gut, liver, and brain BEC, particularly after TNF-α exposure 45 and plays central roles in T-cell immune responses. In mouse colon endothelial cells, MAdCAM-1 expression is increased by TNF-α but not IL-1β, or IFN-γ. 46 Although MAdCAM-1 was weakly expressed on control mouse LEC, it was increased by TNF-α. 46
Cytokines decrease LEC capillary formation
VEGF and cytokines (e.g., IL-18) can be both ‘angiogenic’ and inflammatory.37,56 Despite the decreased cell proliferation in mouse LEC with TNF-α, low TNF-α concentrations did not affect capillary formation, but higher TNF-α concentrations suppressed LEC capillary formation. IL-1β has been reported to increase angiogenesis; 57 our present study found a significant decrease in capillary formation with IL-1β at 10 and 20 ng/ml, (no effect was seen at low levels). Conversely, IFN-γ has been reported as an ‘anti-angiogenic’ factor.33,35,58 We also found that IFN-γ potently reduced capillary tubes in mouse LEC. The rank order of cytokines disturbing LEC capillaries was IFN-γ > IL-1β > TNF-α. Similar cytokines effects were found in human LEC. While TNF-α and IL-1β reduced capillary tube formation, IFN-γ did not. Recently, Eph receptor-ephrin signaling has been linked to lymphatic remodeling;59,60 whether or how cytokines modulate Eph-ephrin signaling to affect lymphangiogenesis needs further investigation. Although inflammation is clearly associated with both increased lymphangiogenesis and elevated tissue cytokines, our results suggest that TNF-α, IL-1β, and IFN-γ may not be potent lymphangiogenic factors in vitro. We have previously reported that inflammation in experimental colitis is associated with a robust increase in lymphangiogenesis (and angiogenesis). 61 Our findings clearly show that these cytokines inhibit lymphatic proliferation and capillary formation, and disturb many lymphatic properties leading to an inflamed phenotype. Therefore an inflamed lymphatic phenotype in vivo likely reflects complex synergic/antagonistic interactions between inflammatory mediators and growth factors that need to be identified and characterized.
Cytokines differentially affect lymphatic TEER
Cytokines such as TNF-α, IL-1β, and IFN-γ disrupt BEC integrity, leading to loss of solute barrier. Inflammation also increases lymphatic drainage; lymphangitis may aggravate clinical and experimental inflammation.62–65 BEC and LEC differential responses to cytokines might also reflect anatomical or species differences.66,67 Disturbances in LEC barrier, especially in 1° lymphatics could increase tissue edema. We found TNF-α and IL-1β decreased mouse and human LEC barrier at 24 h with a rank order in mouse of TNF-α ∼ = IL-1β > IFN-γ; in human LEC the order was: TNF > IL-1β > IFN-γ. IFN-γ did not affect mouse LEC barrier but increased human LEC. In vivo, lymphatic flow is driven by smooth muscle contraction, unidirectional valves, respiratory and skeletal muscle action. At high interstitial pressures, LEC capillaries are pulled open by anchoring emilin-1 and fibrillin filaments, facilitating drainage of fluid, macromolecules, and cells. Cytokine disturbances in clearance could intensify inflammatory disorders (e.g., IBD). It is unknown how LEC barrier changes interact with lymphatic contractility to govern fluid drainage to lymph nodes, but suggest that cytokine dysregulation of LEC barrier will exacerbate inflammation, both by maintenance of an inflamed LEC phenotype and disturbances in tissue architecture.
Conclusions
We describe several alterations in LEC in response to TNF-α, IL-1β, and IFN-γ. LEC ECAM expression to enhance trans-LEC immune cell trafficking or retention may modulate duration or intensity of inflammation. Reduced capillary formation by cytokines suggests that they are antilymphangiogenic at high physiological levels, exception of IFN-γ which decreased capillaries in mouse but has modestly increased in human LEC. Cytokines also dysregulate LEC barrier, leading to diminished fluid clearance and edema. These changes might disturb lymphangion pumping, and combined with altered LEC barrier could severely impair resolution of inflammation.68–71 Our results clearly show cytokines disturb LEC functions in both mouse and human LEC in vitro with differential responses of these two cell types to cytokines.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest or financial ties to disclose.