
Abstract
Abstract
Background:
The lymphatics in the interlobular and subpleural parenchyma contribute to alveolar clearance in the lung, but the information on the remodeling of these lymphatics is quite limited in idiopathic pulmonary fibrosis lungs that contain severe fibrosis in these regions. We compared the alteration of these lymphatics and lymphangiogenesis among idiopathic pulmonary fibrosis and nonfibrotic interstitial pneumonias with a better prognosis.
Methods and Results:
The lung tissue specimens of eighteen patients with idiopathic pulmonary fibrosis (ten surgical biopsies and eight autopsies), six with organizing pneumonia, six with cellular nonspecific interstitial pneumonia, and five normal controls were examined by morphometric analysis of the lymphatics identified by immunohistochemistry. In addition, three-dimensional reconstruction of lymphatics, apoptosis of lymphatic endothelial cells and the cells producing growth factors for lymphangiogenesis were also evaluated. Both the subpleural and the interlobular lymphatics in idiopathic pulmonary fibrosis lungs were significantly decreased in the severe fibroconnective lesions, with rare lymphangiogenesis. The three-dimensional images of the subpleural lymphatics in idiopathic pulmonary fibrosis clearly revealed destruction by fibrosis; apoptosis was observed in these lymphatic endothelial cells. In contrast, organizing pneumonia and cellular nonspecific interstitial pneumonia preserved these lymphatics, and active lymphangiogenesis occurred in the alveolar lesions.
Conclusions:
These results reveal severe damage of the subpleural and interlobular lymphatics in idiopathic pulmonary fibrosis lungs, and suggest impaired alveolar clearance as another pathogenesis of its refractoriness.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a refractory interstitial pneumonia induced by chronic and progressive fibrosis without obvious etiology. 10 We have been interested in lymphatic alterations in the lungs of patients with IPF because of its characteristic distribution of fibrotic lesions in the lungs, normally referred to as usual interstitial pneumonia (UIP). The diagnostic criteria of UIP consist of a heterogeneous distribution of fibrosis, most severely affecting the subpleural and paraseptal parenchyma, 10 where the pleural plexus and interlobular lymphatics are distributed in normal lungs. 1 In addition, we recently observed that fibroconnective tissues had developed in the airway walls of patients who had died of severe asthma, reducing the lymphatic distribution in the airway walls, which was suggested to be a cause of the severe mucosal edema observed in these patients. 11
Although abnormal lymphangiogenesis in IPF lungs has been reported based on the differentiation of alveolar CD11b-positive macrophages into lymphatic endothelial cells, 12 the topographic pattern of lymphatic alteration had not been clearly demonstrated. Therefore, we utilized a morphometric analysis of the subpleural and interlobular lymphatics based on the heterogeneity of fibrotic lesions, as we successfully determined the density of the alveolar capillaries in IPF. 13 In this context, to characterize alterations of the pleural plexus and interlobular lymphatics induced by fibrosis in IPF, we compared these lymphatics among the lung tissues of patients with IPF obtained by surgical biopsy and autopsy, organizing pneumonia and cellular nonspecific interstitial pneumonia (NSIP), in addition to normal control lungs.
Materials and Methods
Clinical samples
The lung tissues obtained by surgical biopsy from 10 patients with IPF (9 males and 1 female, 62 ± 6 years old), 6 with organizing pneumonia (4 males and 2 females, 51 ± 11 years old), 6 with cellular NSIP (3 males and 3 females, 54 ± 6 years old) were fixed in 10% buffered formalin and embedded in paraffin wax. We also examined the autopsy lung tissues from 8 patients with IPF (6 males and 2 females, 63 ± 2 years old) and 5 normal controls without chronic lung diseases (3 males and 2 females, 65 ± 6 years old). Informed consent approved by the ethics committee of Tohoku University was obtained to use these clinical samples for immunohistochemical analysis.
Immunohistochemistry
As a pilot study of immunohistochemistry for lymphatic endothelial cells, we tested four commercially available antibodies: an anti-human podoplanin monoclonal antibody (AngioBio Co., Del Mar, CA), D2-40 (Nichirei, Tokyo, Japan), a rabbit polyclonal antibody against human LYVE-1 (RDI Division of Fitzgerald Industries Intl, Concord, MA), and an anti-human VEGFR-3 goat antibody (R&D Systems, Minneapolis, MN). We also examined three monoclonal antibodies against Prox1, commercially available from AngioBio Co., Abnova, and abcam. We finally chose anti-human podoplanin monoclonal antibody for further study for its specific and higher intense staining. The other monoclonal antibodies used in this study were the antibodies against human CD34 (Nichirei Corporation, Tokyo, Japan) and von Willebrand factor (vWF) (Nichirei). 13 A rabbit polyclonal antibody against human VEGF-C (IBL, Takasaki, Japan) and a goat polyclonal antibody against human VEGF-D (R&D Systems) were also used. The antigen-antibody complex was visualized by Vector Red (Vector Laboratories, Burlingame, CA) and/or diaminobenzidine (DAB), and counterstained by elastica-Goldner stain, a modified elastica-Mason stain. 13 For detecting apoptosis in the tissue sections, an In Situ Apoptosis Detection Kit (Takara Bio Inc., Shiga, Japan) was used. Fluorescence triple immunohistochemistry for lymphatic endothelial cells with anti-podoplanin antibody (in red), for apoptotic cells (in GFP) and for nuclei (in blue) was also examined and the percentages of the lymphatic endothelial cells in apoptosis were compared among lung biopsies obtained from patients with IPF, cellular NSIP, organizing pneumonia, and normal control lungs.
Morphometric evaluation
Morphometric analysis of lymphatic distribution in the lung tissues was performed using an image analyzing system (Lumina Vision, Mitani Corporation, Fukui, Japan). In short, the contours of 50 lymphatics in each case were delineated and the percentages of the lymphatic area in the interlobular or subpleural tissues were calculated. The longitudinal length of each lymphatic vessel was also measured. The distribution density of the new lymphatics developed in alveolar lesions was obtained by counting the new lymphatics per unit area of each tissue section.
Three-dimensional reconstruction images of lymphatics
The three-dimensional digital images of subpleural lymphatics were reconstructed from photographed images at 400 magnification of 25 serial sections of lymphatics, immunostained with the anti-podoplanin antibody, using 3-D image constructing systems (Imaris Ver.3.2/AutoAligner Ver.6.0, Bitplane, Zurich; WinROOF Ver.5.8, Mitani; VGstudio Ver.1.2, Volume Graphics, Heidelberg).
Statistical analysis
For analysis of two unpaired samples, the nonparametric Mann–Whitney U test was used. Significant difference was defined as p < 0.05. All values were represented as the means ± SEM.
Results
Lymphatics in control lungs
In normal control lungs, the lymphatics were distributed in the connective tissue of the visceral pleura, interlobular septa, and the peri-airway or perivascular tissues (Fig. 1). The subpleural lymphatics were observed to connect with interlobular lymphatics, which defined the pleural basis of the lobules (Fig. 1A). Small lymphatics were found at the side of the post-capillary pulmonary veins within the lobules, which were connected to the lymphatics with valves, known as the collecting lymphatics, 1 in the interlobular septa, running alongside the pulmonary veins (Figs. 1B and 1C). The lymphatics beside the pulmonary arteries were well developed so as to come into contact with the alveolar walls (Fig. 1D), described as the juxta-alveolar lymphatics, 14 but no lymphatics existed in the alveolar septa in normal control lungs (Figs. 1A–1D).
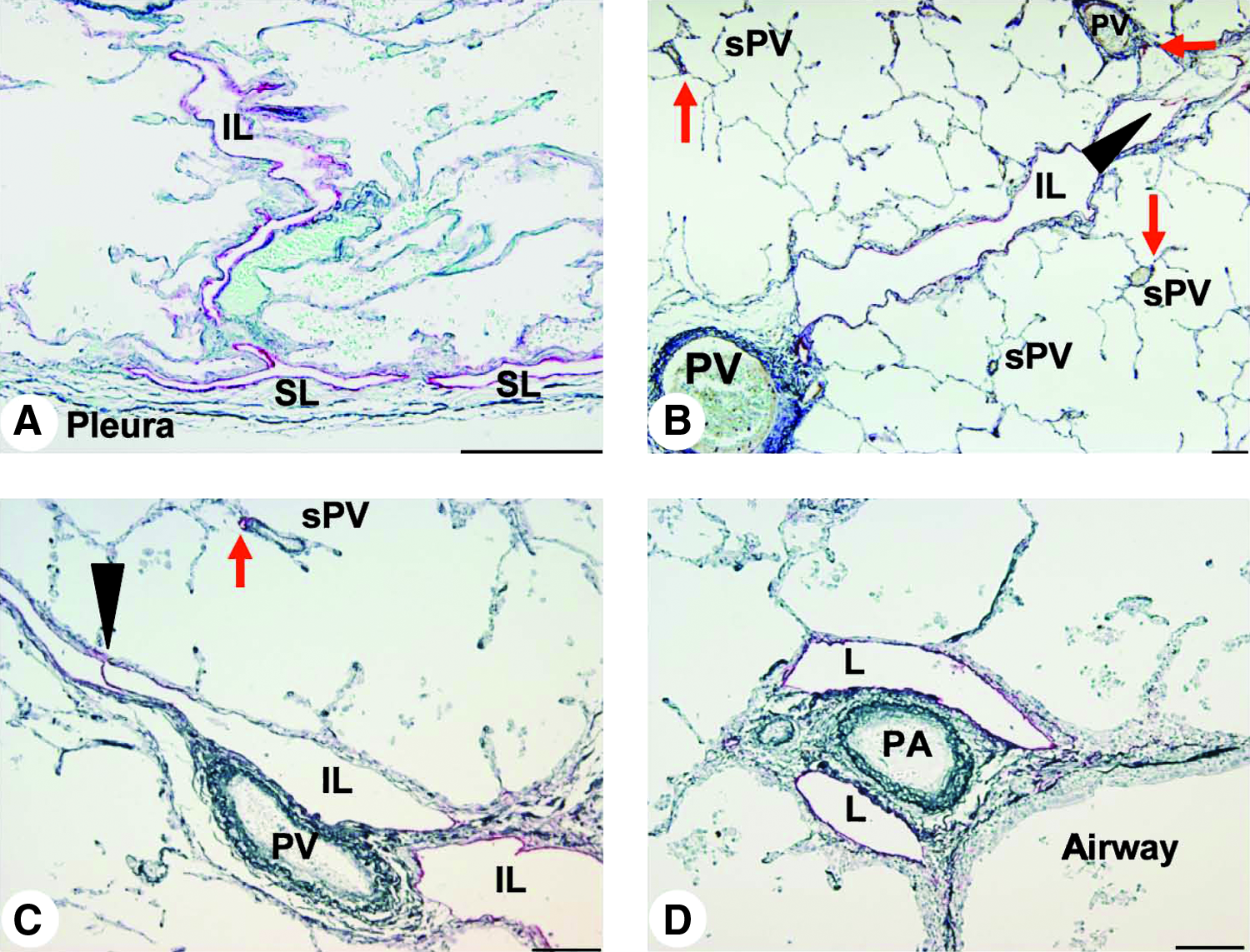
Pulmonary lymphatics in normal control lung. Immunohistochemical staining with a monoclonal anti-podoplanin antibody revealed lymphatic endothelial cells in red, counterstained by elastica-Goldner stain showing elastic fiber in dark blue (scale bar, 100 μm).
Remodeling of lymphatics in interstitial pneumonia
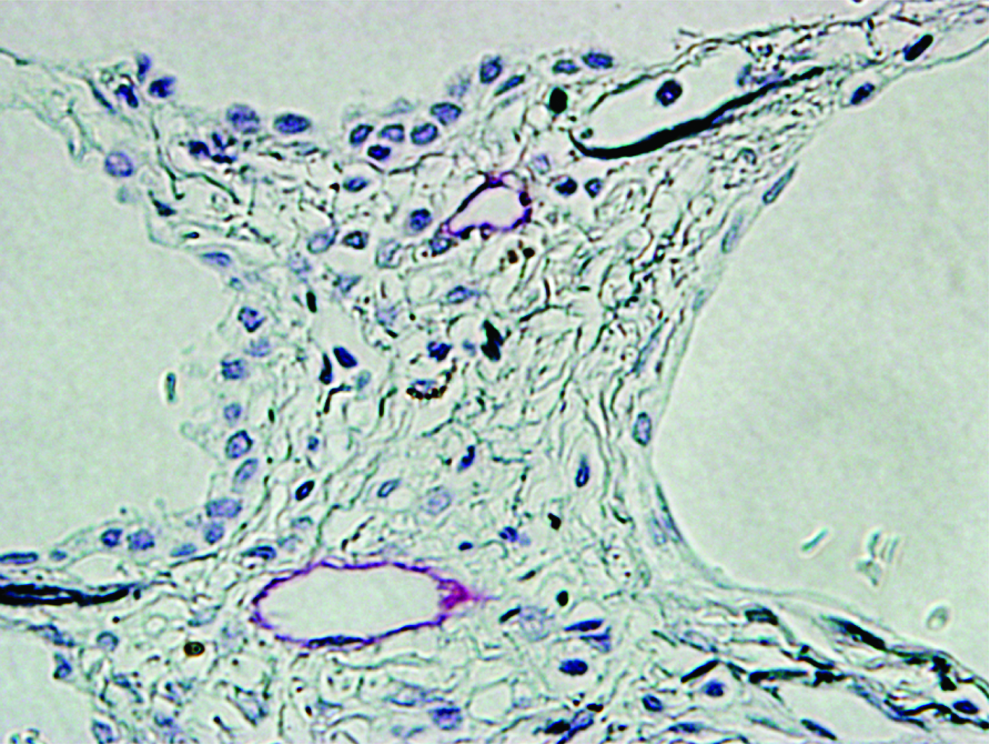
In the lung tissues of the IPF patients examined in this study, which were surgically biopsied or autopsied, severe fibrosis developed in both the subpleural and paraseptal parenchyma. The lymphatics distributed in these regions were remarkably reduced and fragmented (Fig. 2) in comparison with the normal control lungs, as shown in Figure 1. Within the fibroblastic foci, neither lymphatic nor blood capillaries were observed (Figs. 2A and 2C). In contrast, the lymphatics beside the pulmonary arteries where the fibrosis had not increased were sustained without fibrotic damage and a substantial amount of new lymphatics had developed so as to extend from the alveolar lesions (Fig. 2D). To determine whether these fragmented lymphatics were damaged or twisted by the severe fibrosis in these lesions, three-dimensional images of the subpleural lymphatics in the lung tissues of a representative IPF patient were extracted from whole reconstructed tissue images, and compared with those of the normal control lungs (Fig. 3). Contrary to the network-like structures of the subpleural lymphatics in normal control lungs (Fig. 3C), most of the lymphatics distributed in the subpleural fibrotic lesions in IPF were fragmented into pieces (Fig. 3F). We also observed that these disrupted lymphatics in IPF contained apoptotic nuclei (Fig. 4). The percentages of lymphatic endothelial cells in apoptosis in the lungs biopsied from IPF patients were 4.3 ± 0.7% in subpleural lymphatics and 5.3 ± 0.8% in interlobular lymphatics without static difference between them (Fig. 5) However, no apoptotic lymphatic endothelial cells were observed in lungs of patients with cellular NSIP, organizing pneumonia, or normal controls.

Pulmonary lymphatics in IPF lung. Double immunohistochemical staining for podoplanin-positive lymphatic endothelial cells (red) and vWF-positive blood endothelial cells (brown) was counterstained by elastica–Goldner stain (scale bar, 100 μm).
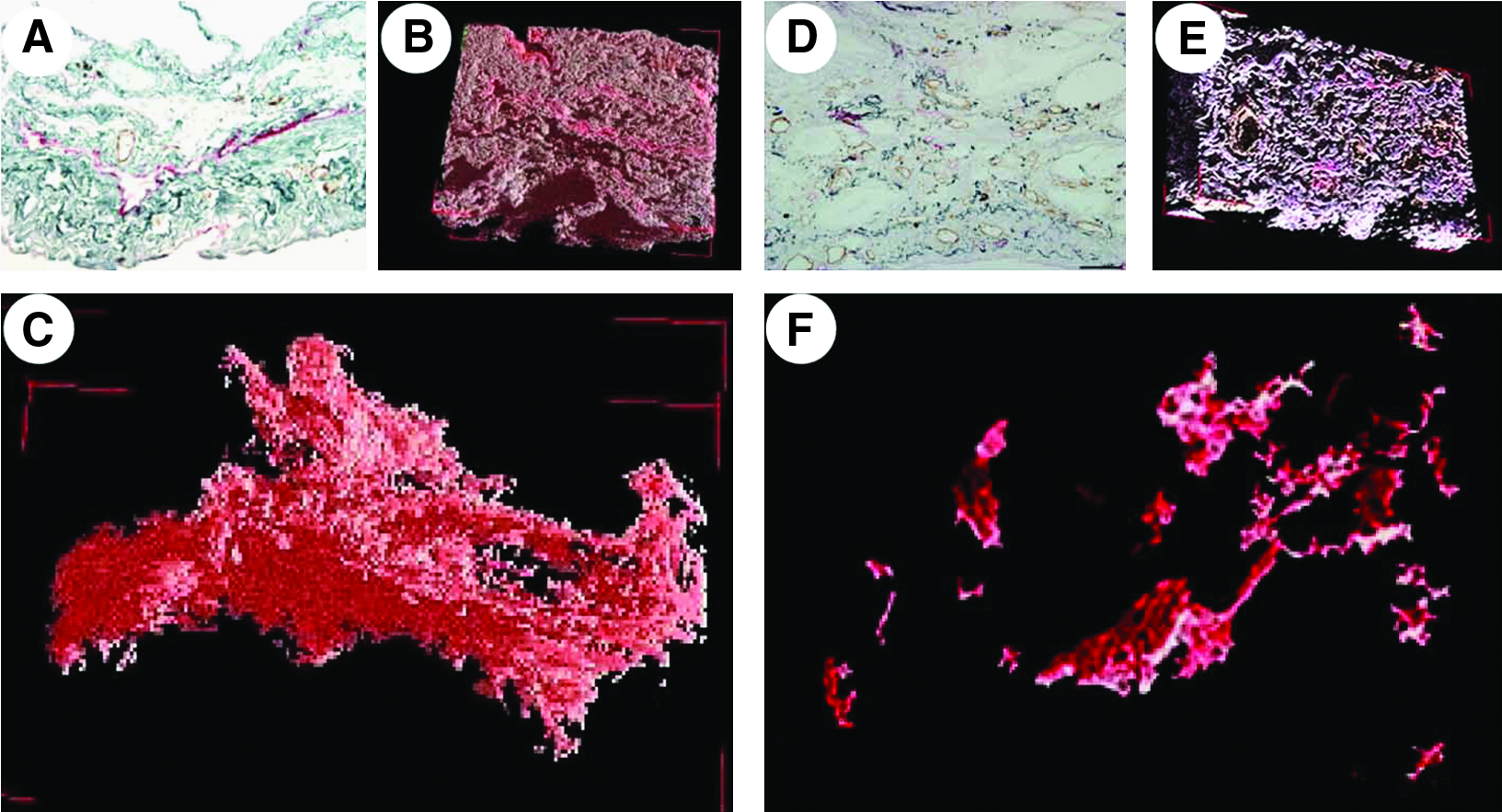
3-D image of subpleural lymphatics in IPF lungs and normal control.
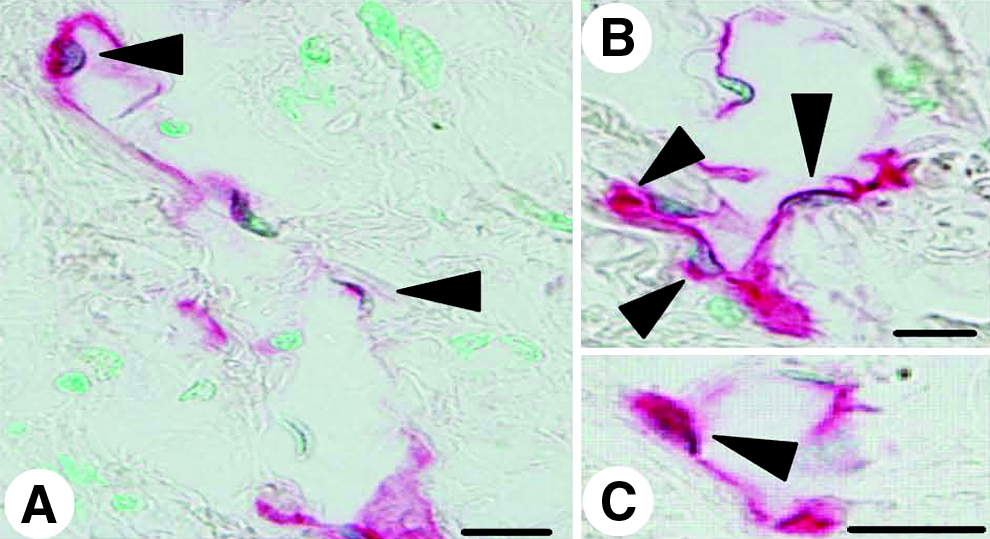
Apoptosis in lymphatic endothelial cells in IPF lungs. Double immunostaining for lymphatics (red) and for apoptotic nuclear cells (dark brown) revealed apoptotic lymphatic endothelial cells (arrow heads) in the fibroconnective tissues of IPF lungs (scale bar, 10μm). The lymphatics containing apoptotic endothelial cells were devastated.
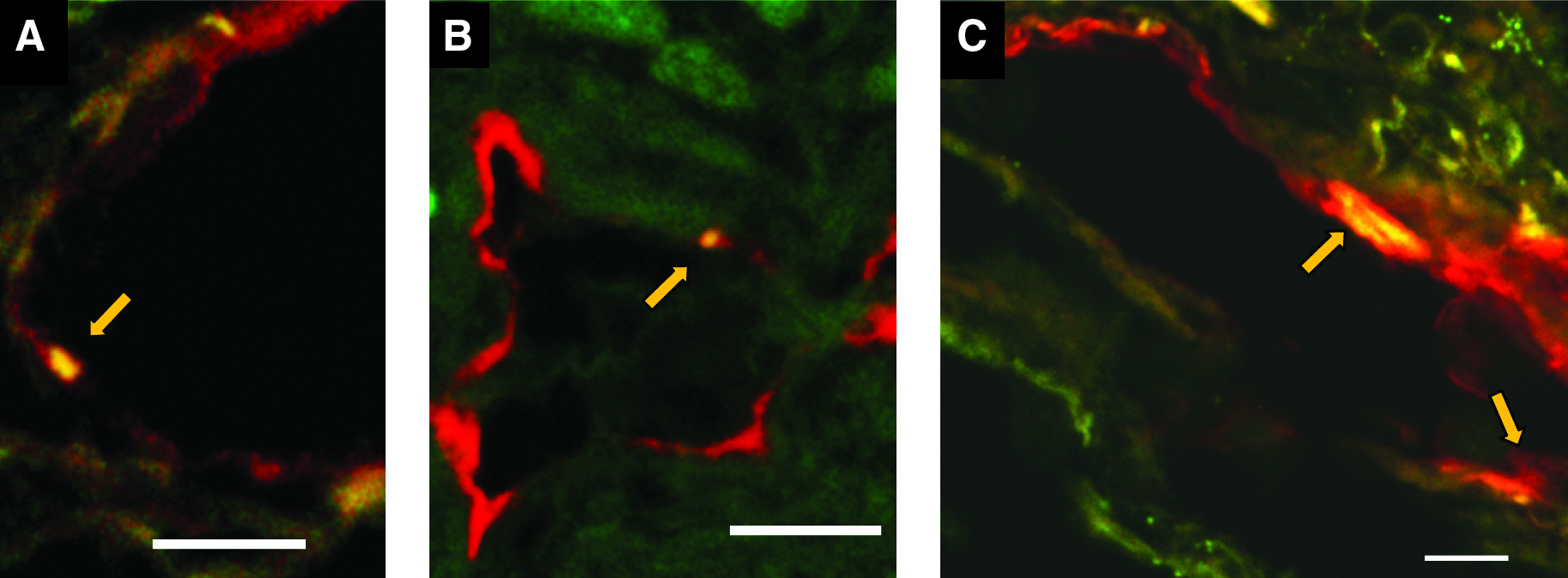
Double immunofluorescence staining for lymphatic endothelial cells and apoptotic cells in IPF lungs. Double immunofluorescence staining for lymphatic endothelial cells (red) and apoptotic cells (green) revealed the apoptotic lymphatic endothelial cell nuclei (merged to yellow, indicated by arrow) which reduced the expression of podoplanin in subpleural lymphatics
In contrast to IPF, both the lungs of patients with organizing pneumonia and cellular NSIP contained the dilated lymphatics in the interlobular septa and subpleural tissues of patients (Fig. 6).
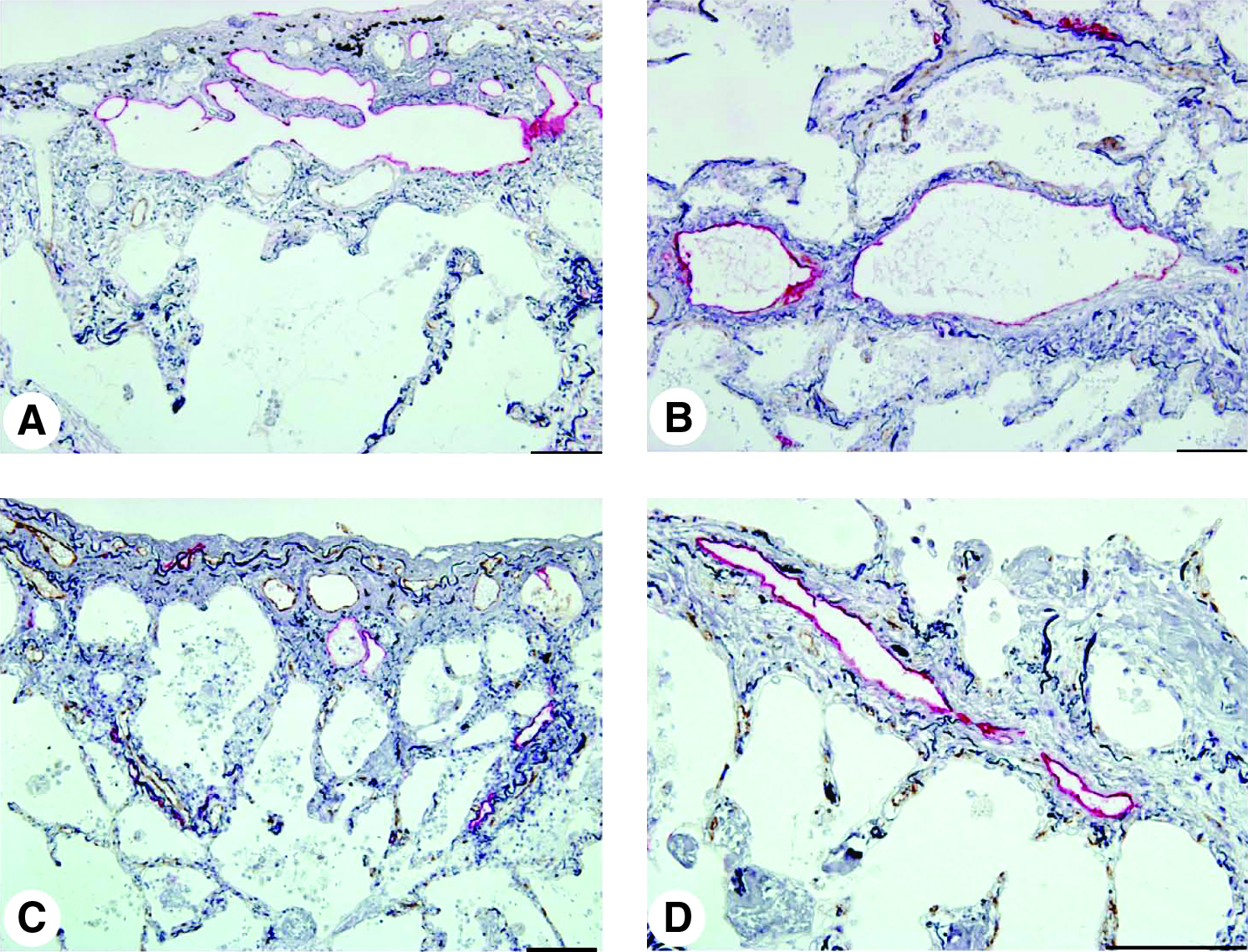
Subpleural and interlobular lymphatics in cellular NSIP and organizing pneumonia. Double immunostaining for podoplanin-positive lymphatic endothelial cells (red) and vWF-positive blood endothelial cells (brown) was counterstained by elastica–Goldner stain (scale bar, 100 μm).
For morphometrical comparison of lymphatic distribution among IPF and the other interstitial pneumonias, we measured the surface area and the length of the lymphatics within the interlobular septa and the subpleural tissues in these lung tissues examined (Fig. 7). The results revealed that both interlobular lymphatics and subpleural lymphatics in the IPF lungs significantly reduced the lymphatic area in comparison with controls, though no significant difference was found among control lungs, cellular NSIP, and organizing pneumonia (Figs. 7A and 7B). When the relative ratios of the interlobular and subpleural lymphatic areas to the surrounding connective tissues were compared among the interstitial pneumonias, both the autopsied and biopsied tissues of patients with IPF, but not cellular NSIP or organizing pneumonia, exhibited reduced lymphatic ratios in comparison with control lungs (Figs. 7C and 7D). These results indicated increased fibrotic tissues had reduced the interlobular and subpleural lymphatic areas in IPF lungs, not only due to shrinkage of these lymphatics. The longitudinal length of lymphatics also revealed the same tendency; it was reduced in both the autopsied and biopsied IPF lungs, but not in cellular NSIP or organizing pneumonia (Figs. 7E and 7F). These results indicated the lymphatics in IPF lungs were fragmented in increased fibroconnective tissues in interlobular and subpleural regions.

Morphometric analysis of interlobular and subpleural lymphatics.
New lymphatics in interstitial pneumonias
In the lungs of cellular NSIP and organizing pneumonia, a substantial number of new lymphatics were frequently observed to extend from the alveolar lesions to existing lymphatics in interlobular or subpleural tissues (Fig. 8). In remarkable contrast, new lymphatics were scarcely distributed in the alveolar lesions in IPF lungs, where these existing subpleural or interlobular lymphatics were enclosed by thick fibrotic tissues (Fig. 9A), although the regenerated alveolar epithelial cells surrounding the fibrotic lesions were immunoreactive for VEGF-C and/or VEGF-D (Fig. 9B and 9C), which were compared with the results of negative control serum (Fig. 10). No lymphatics were observed again within the fibroblastic foci (Fig. 9C).
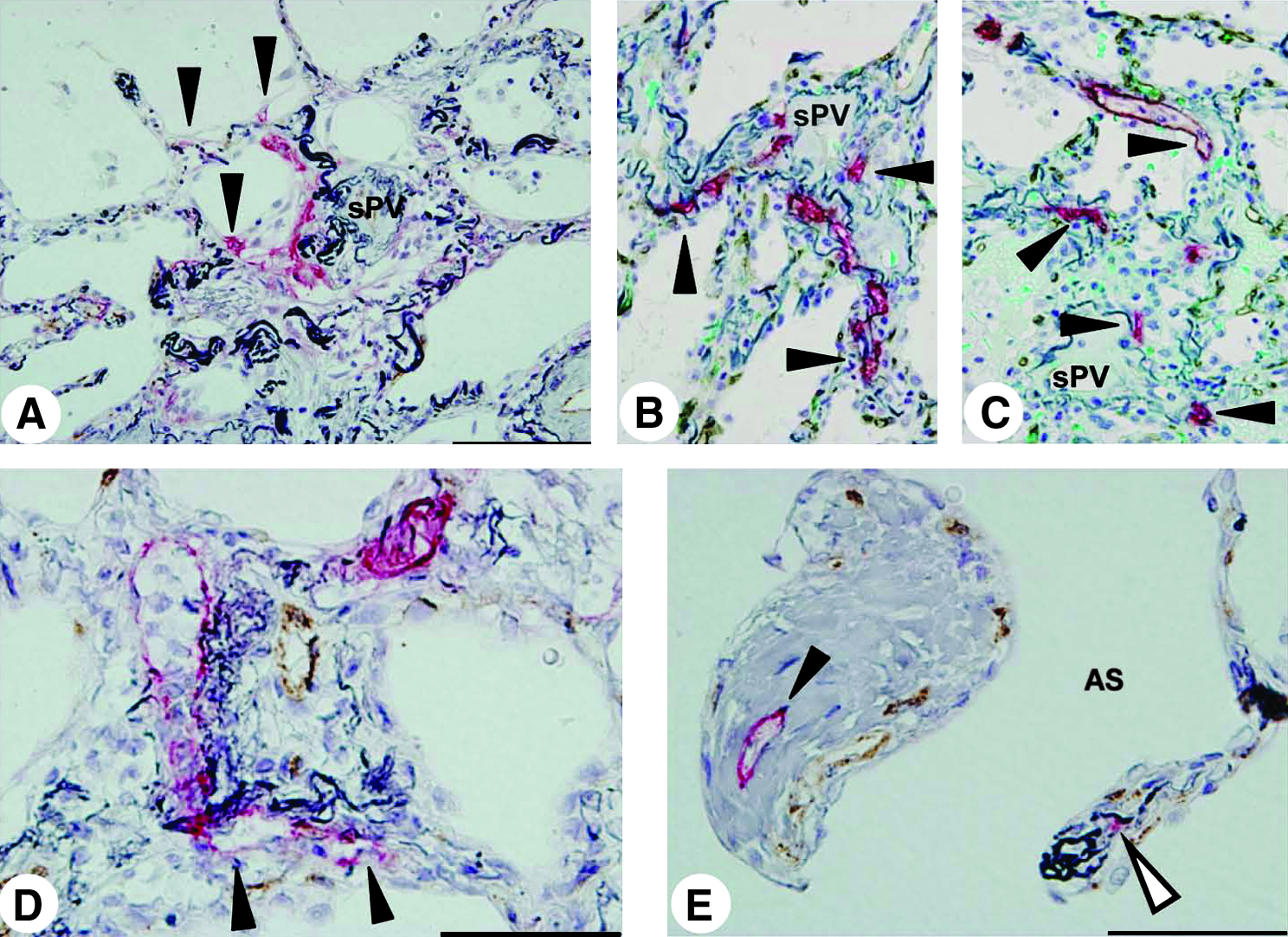
New lymphatic development in cellular NSIP and organizing pneumonia. Double immunohistochemistry of lymphatic endothelial cells in red and CD34-positive or vWF-positive blood endothelial cells in brown, was counterstained by elastica–Goldner stain.
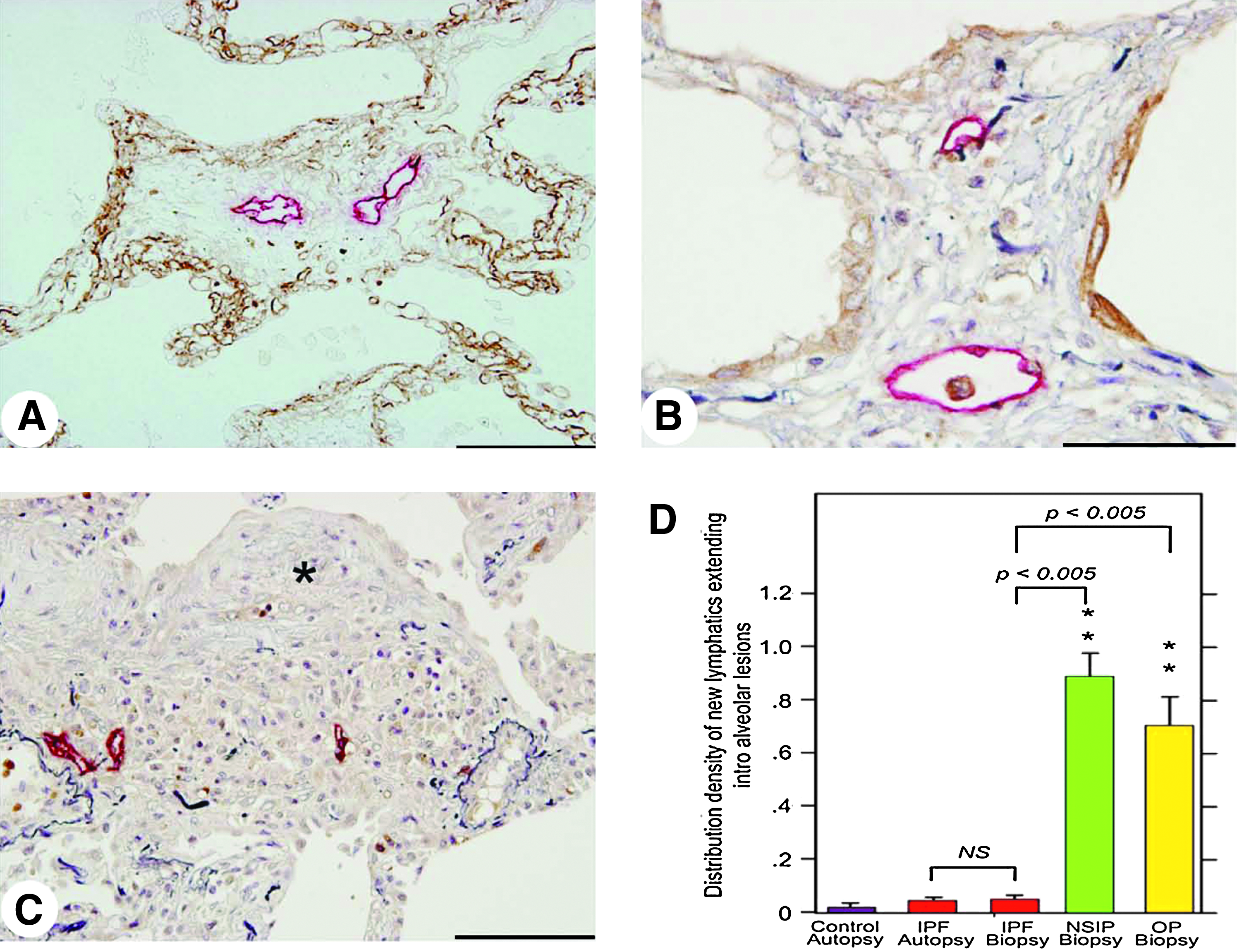
New lymphatics are scarcely developed in IPF lungs.
Double immunohistochemical staining with anti-podoplanin antibody (in red) and with control serum (in brown) to be compared with Fig. 9 (B, C).
To quantify the new lymphatics developed in these tissues, the distribution densities of lymphatics extending into the alveolar space per unit area of lung tissue were calculated. The results clearly revealed a marked increase of the new lymphatics in the alveolar lesions, extending into the existing lymphatics in the lungs of cellar NSIP and organizing pneumonia, but without any significant difference between them (Fig. 9D). No significant increase was found in the lung tissues obtained by biopsy or autopsy from patients with IPF in comparison with normal controls.
Conclusions
To the best of our knowledge, this is the first report of the disappearance of subpleural and interlobular lymphatics in IPF lungs and of a significant decrease of alveolar lymphangiogenesis in comparison with cellar NSIP and organizing pneumonia.
The pulmonary lymphatics are divided into two groups: the lymphatic collecting channels (collecting lymphatics) and lymphatic capillaries. 1 The collecting lymphatics are characterized by the presence of valves to drain the lymph in a specifically directed manner. 15 The walls of the lymphatic capillaries consist of a single layer of endothelial cells surrounded by a very tenuous connective tissue sheath without pericytes, and the endothelium is rendered discontinuous by open cell junctions and intercellular gaps, 1 as is frequently observed in the alveolar lesions of cellular NSIP or organizing pneumonia in this current study.
Our observations of the decrease of lymphangiogenesis in the fibrotic lesions in IPF are supported in part by previous studies on lymphatic alterations in lungs with diffuse alveolar damage.16,17 Yamashita et al. showed that new lymphatics were observed to develop in the proliferative stage, but less in the fibrotic stage in the various exudative, fibrotic and proliferative stages of diffuse alveolar damage in the lung tissues of autopsy subjects. 16 In addition, Mandal et al. reported that the lymphatics in the cases of diffuse alveolar damage were increased only when organizing pneumonia was present, and their survival was better than those with diffuse alveolar damage without organizing pneumonia, including pulmonary fibrosis, which contained less lymphangiogenesis. 17 In contrast, there is a controversial study reporting that the mean area of lymphatics vessels increased significantly with the severity of fibrosis in IPF lungs, 12 which did not, however, clarify the topographic distribution of increased lymphatics or make a comparison with other interstitial pneumonias.
We observed in this study that active lymphangiogenesis occurred in alveolar lesions in cellular NSIP and organizing pneumonia, and also the development of new lymphatics even in IPF lungs, extending from alveolar legions into the lymphatics beside the pulmonary arteries without any evident increase of fibrosis in their airway walls. Considering that the lymphatics in the airway walls are increased in the chronic airway inflammation 8 but significantly decreased in the airway walls with thick fibroconnective tissues of severe asthmatic patients, 10 our observation of poor lymphangiogenesis in fibrotic lesions in IPF lungs may suggest the opposite effects between inflammation and fibrosis on lymphangiogenesis. These observations may be supported by recent evidence that revealed inhibition of lymphangiogenesis by TGF-β1, a key inducer of fibrogenesis.18,19
There are likely to be multifactorial reasons for the disappearance of the lymphatics in fibrotic lesions in IPF. First, the lymphatics in IPF are fragmented and disconnected by massive fibrosis with increased elastic fibers, as shown by the three-dimensional images of the subpleural lymphatics in IPF lungs in this study. Second, lymphatic endothelial cell apoptosis may be induced by the segregation of the epithelial cells and/or alveolar macrophages producing lymphatic endothelial growth factors, VEGF-C or VEGF-D. 20 Because CD45+/CD14+/CD11b+ macrophages in the broncho-alveolar lavage fluid from subjects with IPF form lymphatic-like tube formation in vitro, 12 thick fibroconnective tissues surrounding subpleural and interlobular lymphatics may separate the lymphatic stem cells in the alveolar space from the existing lymphatics, as is observed to a degree in cases of diffuse alveolar damage. 16 Third, the extrinsic driving force of respiration, supporting lymph flow in the lungs, may be diminished in subpleural and interlobular lymphatics in IPF due to restrictive respiratory impairment. 15 In addition, recent new findings on an endogenous inhibitor of lymphangiogenesis18,19,21 may explain a molecular mechanism of disappearance of lymphatics in fibrotic lesions in IPF.
We conclude that the disappearance of the subpleural and interlobular lymphatics in IPF lungs, along with poor lymphangiogenesis, exerts a deleterious influence on IPF by impairing alveolar clearance.
Footnotes
Author Disclosure Statement
Masahito Ebina has received grants from the Ministry of Health, Labour and Welfare, Japan, and from the Japan Society for the Promotion of Science. Kenta Okaya is an employee of Mitani Corporation, Tokyo, Japan. All other authors have no conflicts of interest or financial ties to disclose.
This work was supported in part by a research grant to the Diffuse Pulmonary Disease Research Group by the Ministry of Health, Labour and Welfare of Japan.