
Abstract
Abstract
Background:
Primary lymphedema covers around 10% of all lymphedema cases. Most cases segregate as an autosomal dominant trait and rarely manifest autosomal recessive inheritance. Our research aimed to map and ultimately to hunt the mutation that causes hereditary lymphedema in an extended consanguineous Muslim family consisting of several affected individuals.
Methods and Results:
We attempted molecular diagnosis by applying homozygosity mapping and whole genome linkage analysis. A candidate locus of 2.3 Mb located on chromosome 5q35.3 was identified, yielding an overall LOD score of 3.18. This locus has been previously linked to congenital lymphedema, namely by the FLT4 gene. Mutations in FLT4 that were previously described in Muslim-Israeli families were discarded as culprit using sequence analysis. Sanger sequencing the gene revealed a novel missense variant in exon 28 (NM_182925.4: c.3704C>G; p.Ser1235Cys). This variant has perfect segregation within the extended family and was not previously reported in either common or pathogenic variants databases.
Conclusions:
Our mutation is the first reported pathogenic variant located outside the tyrosine kinase domains of the VEGFR3 receptor, and the second to portray autosomal recessive inheritance. The homozygous substitution of serine by cysteine at position 1235 affects protein tyrosine kinase activity, possibly through a null effect mechanism rather than a negative dominant effect. Our variant is associated with a mild phenotype, possibly reflecting some residual receptor activity, most probably attributed to the variant's location beyond the TK domains.
Introduction
P
The development of the lymphatic system is mediated by a multitude of genes, partakers in the VEGFC/VEGFR3 signaling pathway,9,10 predominantly FLT4 (MIM #136352), FOXC2 (MIM #602402), SOX18 (MIM #601618), and CCBE1 (MIM #612753). 11 Most of the proteins that are encoded by the genes mutated in PL seem to act in a single functional pathway involving VEGFR3 signaling.2,9,10
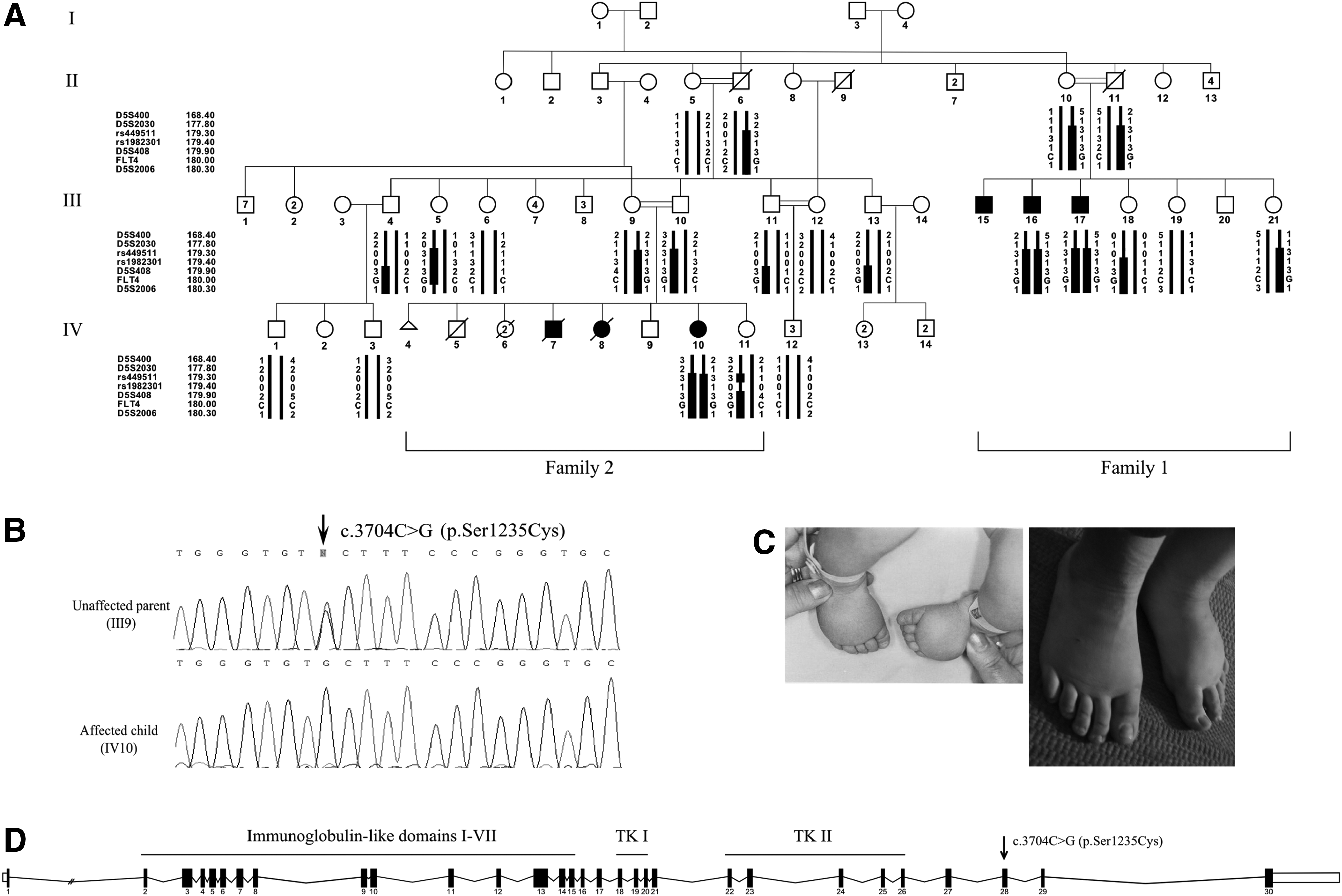
Here we report an extended inbred kindred of Muslim Israeli–Arab origin, with autosomal recessive congenital lymphedema (Fig. 1A; Table 1).
All individuals were born at term; NVD, normal vaginal delivery;
Site of edema: +unilateral; ++ bilateral; +/− most probably bilateral.
Edema severity: +mild; ++ moderate; +++severe.
Methods
Participants and families
The studied kindred embraces two families, each presenting with three affected siblings (F1: III15, III16, III17, and F2: IV7, IV8, IV10, respectively), born to healthy first-cousin parents.
Blood samples were drawn from both healthy (n=17) and affected (n=3) participants for molecular analysis. The study was approved by the Helsinki committee at Rambam Health Care Campus, Haifa, Israel and performed in accordance with the ethical standards on experiments on human subjects. Signed informed consent (self and parental) was obtained as customary.
Molecular analysis
We undertook homozygosity mapping and linkage analysis by a 6000 single nucleotide polymorphisms (SNPs) microarray, on 12 family members, both affected (n=3) and healthy (n=9), using Illumina's (San Diego, CA, USA) HumanLinkage-12 BeadChip. Genotyping results were scanned for runs of homozygosity in affected individuals and were compared to known lymphedema-associated regions. Analysis with polymorphic microsatellite markers, namely D5S400, D5S2030, D5S408, and D5S2006, was used to reinforce the linkage to the candidate region on chromosome 5q35.3. Haplotypes were manually constructed and LOD scores were calculated using Superlink Online v1.5 software (http://bioinfo.cs.technion.ac.il/superlink-online/). The coding regions and flanking intron–exon boundaries of FLT4, the candidate gene within the homozygous region, were Sanger sequenced, starting with exons known to harbor pathogenic mutations and followed by the rest (primer sequences are available on request). A causative mutation, NM_182925.4: c.3704C>G; p.Ser1235Cys, was detected in exon 28, and was confirmed by a PCR-RFLP assay developed by us, using the following primers: FLT4_28F 5′ GAAGTTCGAGGAGGGTTGC 3′ and FLT4_28R 5′ GCAGCGTCAGGAATTCGTA 3′. The C to G substitution abolishes the native site for the BseLI restriction enzyme, yielding DNA fragments of 252 bp and 65 bp in homozygous individuals, and 152 bp, 100 bp, and 65 bp in wild-type samples.
Whole-exome analysis was executed using the HiSeq™ 2000 sequencer (Illumina), on genomic DNA of an affected individual (Fig. 1A, III16), in order to probe lymphedema-related and novel genes for putative variants. Exome enrichment was done using Illumina's TruSeq® exome enrichment kit, and alignment and mapping of the reads to the reference genome (build GRCh37.64) were done using BWA (Burrows-Wheeler Aligner) v0.6.1 and SAMtools v1.1.18 software.
A population screening for the c.3704C>G; p.Ser1235Cys mutation in FLT4, was conducted among 100 unrelated Muslim Israeli–Arab individuals.
Total RNA was extracted from lymphocyte cell-lines of both affected (n=1) and healthy (n=5) family members, using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed into cDNA by the QuantiTect reverse transcription kit (Qiagen) using RT primer mix according to the manufacturer's protocol. The cDNA was used to quantify the expression of wild-type and mutated FLT4 in comparison to the expression of the housekeeping gene ACTB (Actin-beta) (MIM #102630), using real-time PCR ΔΔCT analysis by Rotor gene Q series software (Qiagen).
Results
Clinical evaluation was available for all participants, as depicted and summarized in Table 1.
Family 1: the affected males (n=3) presented shortly after birth with severe bilateral edema of the lower extremities and hydrocele testis, necessitating recurrent surgical drainage. Other findings included walking difficulties, pain, leakage of serous fluid via the toes, and scaring and discoloration of the skin due to recurrent events of cellulites.
Family 2: the parents recall numerous spontaneous abortions and two newborns deceased at 2–3 days of age. Individual IV7, male, was born with severe, most probably bilateral, edema of the lower extremities and died at the age of 2 days, presumably from a severe heart defect. IV8, a female, was born with severe, most probably bilateral, edema of the upper and lower extremities and died at the age of 3 days, seemingly due to a severe heart defect. IV10 is the only affected living sib in family 2. She presented at birth with moderate bilateral edema of the lower extremities (Fig. 1C). No discomfort or walking difficulties were reported. A significant decrease in the severity of the edema was observed over the years (Fig. 1C).
Whole-genome homozygosity mapping revealed a 2.3 Mb candidate locus on chromosome 5q35.3, with an overall highest LOD score of 2.4. Analysis with the polymorphic microsatellite markers reinforced the linkage to this region, yielding a LOD score of 3.18 for D5S408. The locus encloses the FLT4 gene, majorly implicated in autosomal dominant Milroy's disease.12,13 Sanger sequencing excluded mutations in exons known to harbor pathogenic variants. A novel missense mutation in exon 28, NM_182925.4: c.3704C>G; p.Ser1235Cys, was identified (Fig. 1B, D). Haplotypes depicting the segregation of markers, SNPs, and the disease allele, within the family, are portrayed in Figure 1A. This variant has not been recorded in the common databases (e.g., dbSNP, 1000Genomes, Exome Variant Server). Evolutionary conservation analysis by UniProt (http://www.uniprot.org/) shows that the amino acid serine in position 1235 of the protein is conserved in different species, and both PolyPhen-2 v2.2.2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/) predicted this variant to be damaging. The variant segregated as expected among family members and was not observed among 100 unrelated Muslim Israeli–Arab individuals screened for the mutation.
To exclude other genetic variants that may explain the clinical presentation of our patients, whole-exome sequencing (WES) was performed on an affected individual (Fig. 1A, III16). The first step in the WES analysis probed for novel variation in lymphedema-related genes, such as FLT4, FOXC2, SOX18, CCBE1, KIF11 (MIM #152950), TSC1 (MIM #191100), PTPN11 (MIM #163950), GLA (MIM #301500), PEPD (MIM #170100), LCS1 (MIM #214900), MPI (MIM #602579), and FAT4 ((MIM #612411)). No homozygous novel variants other than FLT4 c.3704C>G; p.Ser1235Cys, described above, passed filtration. To further verify that our phenotype is not caused by a new lymphedema gene, the next analytic step explored all novel homozygous variants obtained by WES (n=56). Only one missense variant, NM_001528.2: c.275C>G; p.Ser92Trp (rs199767749) in the HGFAC gene (MIM #604552) could be associated with lymphatic disruption as it serves as a HGF (hepatocyte growth factor) activator. 14 However, this variant did not segregate within the studied family.
Real-time PCR analysis yielded no expression of FLT4, in either healthy or affected individuals, compared to the reference gene, ACTB (MIM #102630). This reinforced the notion that the gene is majorly expressed in lymphatic endothelial cells.
Discussion
Here we describe a novel missense mutation in the FLT4 gene (NM_182925.4: c.3704C>G; p.Ser1235Cys), the first reported pathogenic mutation located outside the tyrosine kinase (TK) domains of the VEGFR3 receptor. Our family is the second reported to display autosomal recessive inheritance associated with Milroy disease. Milroy disease most commonly presents with bilateral lymphedema of the lower extremities and hydrocele testis. 15 This phenotypic expression is consistent with most of our patients. Nevertheless, cases of atypical clinical presentation of hereditary lymphedema type I had been previously described. 16 In our extensive family, atypical signs and symptoms are also present, raising the possibility of another lymphedema-related syndrome. Milroy disease is known to associate with mutations in the FLT4 gene, which explain ∼70% of cases. 7 Hereby provided genetic evidence, which comprises of homozygosity mapping, segregation analysis, population screening, and exclusion of other variation by WES, places our novel FLT4 variant as causative of the studied phenotype.
The FLT4 gene contains 30 exons and encodes a protein with an extracellular region that embraces seven immunoglobulin-like repeat domains, a trans-membrane region, and two intra-cellular TK domains (TK-I and TK-II) (Fig. 1D). The encoded protein is a vascular endothelial growth factor receptor (VEGFR3), which regulates the development and maintenance of the lymphatic system (lymphangiogenesis). 19 VEGFR3 is activated by two ligands: the vascular endothelial growth factor C (VEGFC, MIM #601528) and the vascular endothelial growth factor D (VEGFD), resulting in homodimerization or interaction with VEGFR2 (MIM #191306) to form a heterodimer.20–23 Activation of VEGFR3 leads to generation of downstream signaling docking sites and results in regulation of proliferation, migration, and survival of lymphatic endothelial cells. Disruption of VEGFR3 in mice hinders the development of the vascular and the lymphatic network, and leads to embryonic death due to defective development of lymphatic vessels, large blood vessels, and cardiovascular failure.24,25
To date, 67 mutations in the FLT4 gene have been reported.18,23 Most are missense mutations; insertions and deletions were occasionally reported. All of the reported mutations are located within the TK domains of the protein, thereby causing a sufficient reduction of tyrosine-kinase activity and limited lymphatic development. All described mutations in this gene, but one, are inherited as autosomal dominant traits.8,16,23 The single functional wild-type allele of VEGFR3 is necessary for primary vasculature and viability. In contrast, a single case of recessive inheritance, caused by a novel homozygous mutation, a transition from alanine-to-threonine in amino acid 855, located in the ATP binding domain of the VEGFR3 receptor was reported by Ghalamkarpour et al. 8 Our family is the second to describe primary vascular development in patients with a homozygous VEGFR3 mutation; however, a dysfunctional lymphatic network results in peripheral lymphedema.
Expression studies have shown that mutations in FLT4 lead to inactivation of the TK domains, thereby failing signal transmission by the receptor and downstream activation. 26 Karkkainen et al., 17 established that, irrespective of the fact that mutations in FLT4 block TK activity, the product of the wild-type allele can transphophorylate the mutant receptor to some extent. This allows partial receptor function, associated with a lymphedema phenotype in autosomal dominant mutation carriers.
Functional studies can provide a comprehensive understanding of the pathogenic effect of the mutant allele c.3704C>G; p.Ser1235Cys. However, in adults FLT4 is expressed exclusively by lymphatic endothelial cells,5,20 thereby restricting the availability of biologic materials for further functional studies. Dixelius et al., 27 described six tyrosine residues located beyond the TK domains of the protein, which serve as phosphorylation sites. Two of these, namely Tyr-1230 and −1231, flank our mutated variant. In addition, limited serine phosphorylation was observed in vitro, coinciding with the fact that the amino acid serine can undergo phosphorylation. In our case, the homozygous substitution of serine by cysteine at position 1235 affects protein tyrosine kinase activity, possibly through a null effect mechanism rather than a negative dominant effect. Our variant is associated with a mild phenotype, possibly reflecting some residual receptor activity, most probably attributed to the variant's location beyond the TK domains.
Footnotes
Author Disclosure Statement
The authors declare no conflict of interest.