
Abstract
Background:
Complex lymphatic anomalies (CLA) are a group of conditions that pose diagnostic and therapeutic challenges due to their rarity and overlapping clinical findings. This case series describes the complex pathology and novel combination therapies of three patients diagnosed with various types of CLA.
Methods and Results:
A retrospective review of medical records was performed for three patients treated for CLA between 2011 and 2019. Diagnostics, imaging, treatment, and follow-up were reviewed in the electronic medical record and combined with the literature review within the analysis. One patient had involvement of her skull base and ear canals, diagnosed after ear canal abnormalities were detected on computed tomography following meningitis. The second patient had involvement of her posterior ribs and T7–T12 vertebral bodies, with thoracic instability requiring a back brace. The third patient had involvement of his left lower extremity and hemipelvis, necessitating a left above the knee amputation. Case 1 progressed on sirolimus and pamidronate but responded to zoledronic acid (ZA). She developed flares of coagulopathy and cellulitis that required reinforcement with vincristine and steroid pulses. Similarly, case 2 progressed on sirolimus and ZA alone, but achieved stable disease with added vincristine. Upon further disease progression, stabilization was obtained by the reinforcement of ZA. Case 3 required a combination of surgery as well as medical management with sirolimus and pamidronate. All three patients now have stable disease.
Conclusion:
This case series depicts a multidisciplinary and multiagent approach to the management of CLA with severe bony involvement using sirolimus, bisphosphonates, vincristine, and steroids.
Introduction
Complex lymphatic anomalies (CLA) is a term that has emerged recently to describe a group of medical conditions with overlapping diagnostic features and clinical findings, namely generalized lymphatic anomaly (GLA), Gorham–Stout disease (GSD), and kaposiform lymphangiomatosis (KLA).
GLA is characterized by diffuse or focal proliferation of dilated lymph vessels affecting the bone, liver, spleen, mediastinum, lungs, and soft tissues. Thoracic involvement is typically associated with poor prognosis when compared with bone or soft tissue lesions. 1
KLA is a subtype of GLA and often presents with an aggressive course, complicated by the development of consumptive coagulopathy and thrombocytopenia along with the typical bony involvement seen in GLA. It has a distinctive histopathologic finding of tight clusters of spindle cells with hemosiderin deposition that is not seen in the other types of CLA.
GSD, also known as massive osteolysis or vanishing bone disease, is an idiopathic disorder in which patients undergo bone resorption without associated proliferation, often with significant soft tissue swelling. A characteristic finding in these patients is the spontaneous and progressive proliferation of blood and lymph vessels that gradually replace the bone and marrow space. 2 First noted in 1838 by Jackson as a case of vanishing bone disease, it was further described in 1955 by Gorham and Stout. 3 Building on Gorham and Stout's observation that affected bones contain innumerable dilated endothelium-lined vessels, subsequent investigators reported these channels to represent blood vessels and/or lymphatic vessels. 4 The emergence of immunohistochemical markers, in particular, LYVE-1 and D2-40, helped us in further appreciating the presence of lymphatic vessels.4,5
The clinical presentation for these patients can be quite variable and typically depends on the part of the skeleton involved. Some cases are indolent, but others like those described in our series may experience rapid onset of symptoms associated with pain, infection, organ dysfunction, or difficulty in breathing secondary to chylothorax. 4
Treatment for CLA has involved medications, surgery, and even radiation in isolated cases; however, no consensus regarding treatment exists. 6 Given that vascular malformations harbor mutations involving vascular endothelial growth factor (VEGF) and the phosphoinositide-3-kinase pathway (PI3K), sirolimus has gained momentum as the drug of choice in slowing the progression of CLA due to its ability to inhibit the mammalian target of rapamycin (mTOR), a kinase involved in the PI3K pathway.7,8 Based on the results of this case series, we propose that another important medication to consider in these patients includes bisphosphonates such as pamidronate and zoledronic acid (ZA) due to their anti-osteoclastic and anti-VEGF properties, which are pathways that have previously been reported to be overactive in GSD and possibly in all CLAs.9,10
In this case series, we discuss our approach to the management of three patients with CLA, specifically highlighting the diagnostic challenges involved in these cases along with a unique multiagent approach, which utilized a novel combination of sirolimus, steroids, bisphosphonates, and vincristine.
Materials and Methods
This case series consists of a retrospective review of medical records for three patients treated for CLA between 2011 and 2019. Diagnostics, imaging, treatment, and follow-up for each patient were reviewed in the electronic medical record and combined with the literature review within the analysis. This case series was waived of institutional review board approval per institutional policy since it did not include greater than three patients.
Results
Case 1
A 12-month-old Hispanic female who presented in June 2014 with pneumococcal meningitis and bacteremia was subsequently found to have hearing loss with markedly widespread abnormalities in ear canals on computed tomography (CT). Magnetic resonance imaging (MRI) revealed herniated cerebellar tissue bilaterally, along with infiltrative lesions in the skull base and cervical vertebral bodies. Biopsy of mastoid bone showed extensive osteolysis and marrow replacement by dilated lymphatic channels, highlighted by D2-40 immunohistochemical stain. These findings combined strongly suggested GSD. She started sirolimus and bisphosphonate therapy with pamidronate. MRI in October 2014 showed progressive disease, prompting a switch from pamidronate to ZA, which was stopped in 2016 due to radiographically evident stable bony disease, while sirolimus was continued.
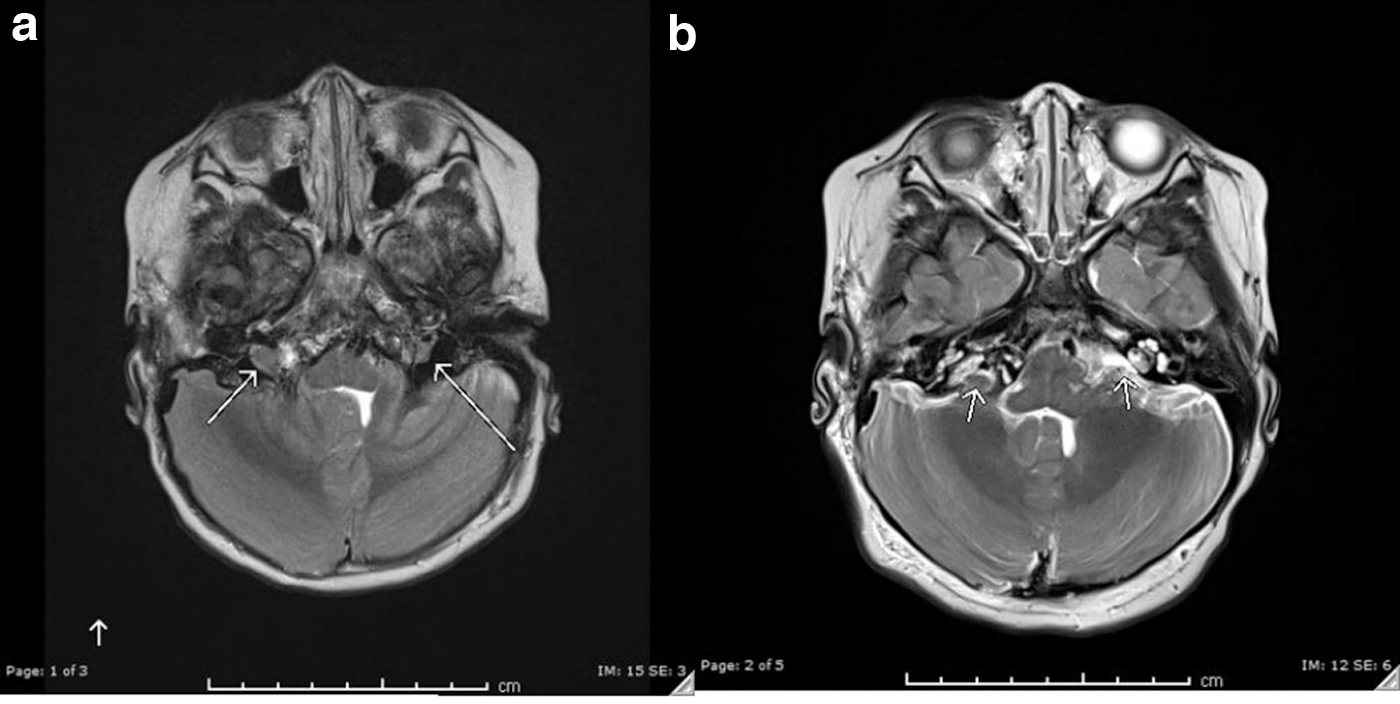
Subsequently, she presented twice with facial swelling, redness, and pain along with consumptive coagulopathy. Laboratory results at the time were remarkable for D-dimer >20 (0–0.49 fibrinogen equivalent units), fibrinogen 153 (200–400 mg/dL), prothrombin time 15 seconds (11.4–15.1 seconds) and partial thromboplastin time 32 seconds (22.4–37.5 seconds), and platelets of 41,000 (150,000–450,000/μL). During the same admission, biopsy of a soft tissue neck mass showed distended D2-40 immunoreactive lymphatic channels coursing through the soft tissue and around a reactive lymph node. Interestingly, none of the pathological features of KLA were seen on biopsy despite the presence of coagulopathy. Based on the abnormal laboratory values and clinical symptoms, she was then treated with weekly vincristine, steroid pulses, and ZA while continuing sirolimus. Early attempts to space out vincristine and discontinue the steroids led to further redness and swelling, after which both medications were resumed. Eventually, she was weaned off vincristine and managed only with ZA and short pulses of steroid to control presumed flares of her disease. Her most recent flare required only a short steroid pulse without further vincristine treatment. Currently, she is successfully maintained on once yearly ZA and daily sirolimus. Follow-up MRI in January 2019 showed significant improvement in both bony and soft tissue findings (Fig. 1a, b).
Case 2
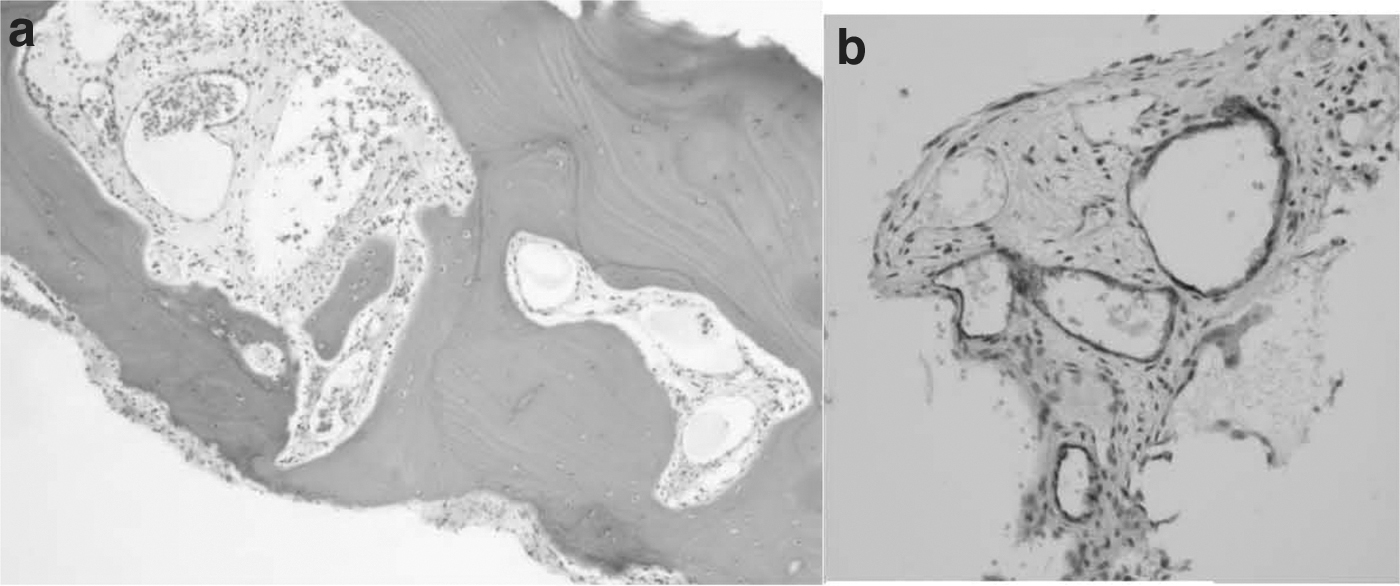
A 9-year-old Hispanic female initially presented at an outside hospital in September 2012 with back pain and shortness of breath. Chest X-ray demonstrated bilateral pleural effusions (PLeff) and suspicious lytic lesions in the spine (Fig. 2a). She was then transferred to our institution for further care. Pleurocentesis demonstrated bilateral chylous PLeffs, which showed only benign lymphocytes on cytology. CT demonstrated extensive lytic disease of T7–T12 thoracic vertebrae. Biopsies of the posterior ninth rib and a thoracic spinous process displayed erosion of the bone with an abnormal lymphatic vascular proliferation, highlighted by D2-40 immunohistochemical stain (Fig. 3a, b). This conferred a preliminary diagnosis of GSD, although this diagnosis was later deemed to better fit GLA due to the fact that GSD rarely leads to PLeff and the bone lesions were separate, discrete lesions rather than the contiguous lesions seen in GSD. This patient was experiencing severe thoracic instability due to the many lesions in her vertebrae, and she required wearing a brace at all times except when lying supine. Bilateral chest tubes were placed for 2 months due to persistent and severe PLeffs. Her right chest tube was removed successfully; however, she continued to have persistent reaccumulation of the left PLeff. After 3 months, she was discharged home with a left chest tube still in place, which was successfully removed 1 month later.

Upon diagnosis, she received daily oral sirolimus and monthly intravenous (IV) ZA. After the first month, the bilateral PLeffs and chylous drainage persisted, prompting the addition of weekly IV vincristine. Imaging in November 2012 showed stable bony disease, and by January 2013, both chest tubes were discontinued following a CT that showed improving bony disease with stable small PLeff (Fig. 2b).
Two years later, she experienced an increase in the size of her previously stable PLeff, prompting the addition of once monthly ZA for 3 months, after which the same dose of ZA was given once every 3 months based on expert recommendation. In September 2016, clinically and radiographically evident improvement of her bony disease led to discontinuation of her brace. Clinically, she is now doing well and remains without a brace. She has mild scoliosis, and her scans continue to show improvement. Her ZA has been decreased to once every 6 months, and she continues on oral sirolimus.
Case 3
A 2-year-old Hispanic male initially presented to a facility in a foreign country for a left leg mass and subsequently underwent resection of a presumed osteoid osteoma from his left lower extremity (LLE). After several more surgeries, he was diagnosed with chronic osteomyelitis. In 2011, at 5 years, he had a bone growth stimulator placed and was diagnosed with GSD at that time. In May 2013, at 7 years, he presented to the emergency department of our institution for the first time with fever, LLE pain, and swelling. He required surgical debridement, removal of his bone stimulator, and treatment with IV antibiotic for Staphylococcus aureus osteomyelitis. At the time, he required a walker to ambulate, had a leg length discrepancy of almost one foot, and had bilateral chronic lymphedema up to the knees. In May 2013, medical management was initiated with combination of IV pamidronate and sirolimus. His left lower leg was amputated in June 2014, after which he remained in remission for 2 years.
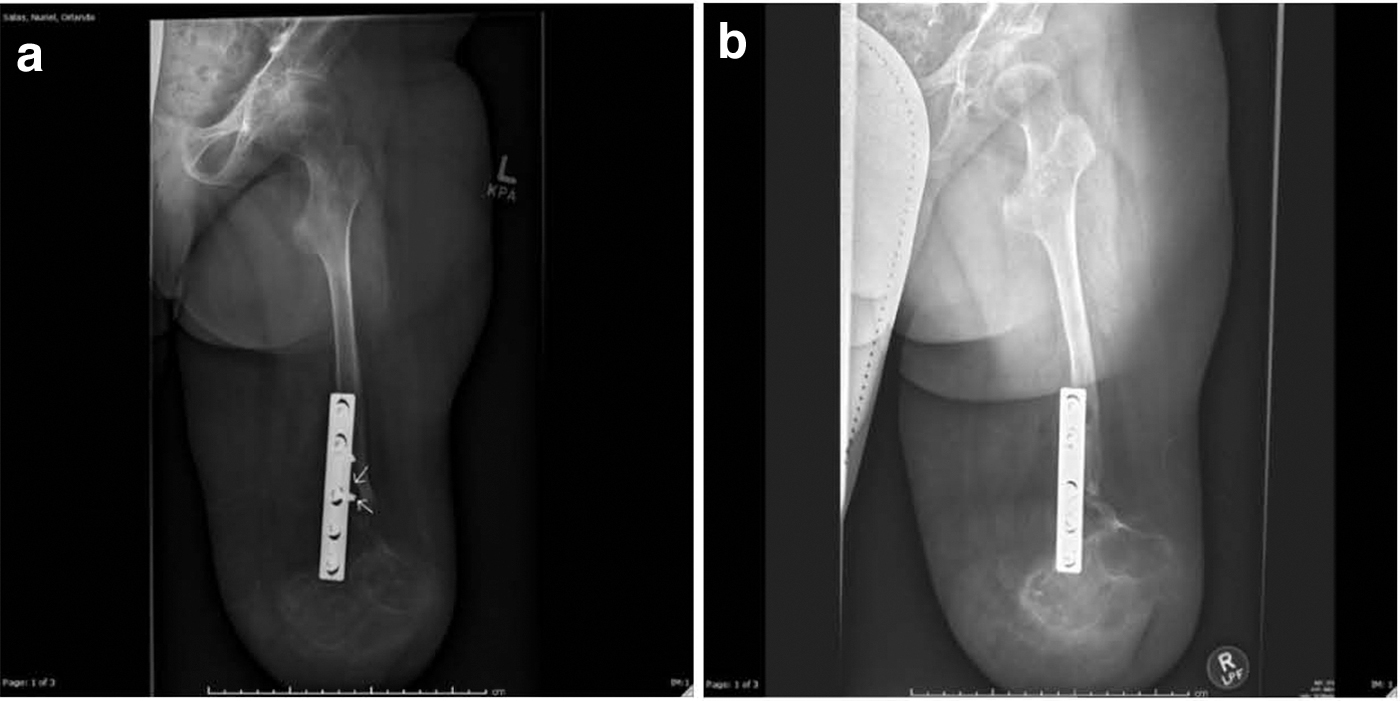
In June 2016, X-rays showed increased lucency around his surgical hardware concerning for progression of GSD (Fig. 4a). Concurrently, his sirolimus level was subtherapeutic; however, this was suspected to be due to noncompliance secondary to communication barrier between providers and family. This was addressed appropriately, and the most recent X-ray in 2018 showed stabilization of bony disease compared with films from 1 year prior (Fig. 4b). He continues on oral sirolimus therapy and is ambulatory with his prosthesis.
The side effects of sirolimus observed in these patients were limited to hypertriglyceridemia and mucositis, which were both managed without necessitating any changes to treatment course. Fortunately, no serious adverse effects were noted with the other agents. Patient summaries are depicted in Table 1.
Patient Summaries
CLA, complex lymphatic anomalies.
Discussion
The various subtypes of CLA—including GSD, GLA, and KLA—present with many diagnostic and therapeutic challenges due to common, nonspecific clinical findings and overlapping patterns.
Gorham–Stout disease
GSD is a rare sporadic condition of unknown origin with potentially devastating consequences. The clinical presentation can be variable, depending on the bone(s) involved, which may be single or multiple contiguous. Localized pain is the most common symptom, and patients may also present with weakness, swelling, or pathological fracture, depending on the bones involved, or dyspnea secondary to chylothorax. 4 The shoulder, pelvis, spine, and ribs are the most common sites of involvement in GSD. The affected bones have been reported to be associated with abnormal proliferation of lymphatic vessels rather than blood vessels, along with resorption of bone without increased activity of osteoblasts. 11
Because of the relative rarity of the condition and nonspecific presenting features, the differential diagnosis at presentation is quite broad. 12 This often leads to delay in diagnosis as well as significant morbidity secondary to complications. Biopsy samples are expected to display a positive D2-40 immunohistochemical stain, highlighting the abnormal lymphatic channels. Initial laboratory studies are rarely helpful, and early imaging will show a patchy osteoporosis-like picture; concentric bone loss will be seen later in the disease process. Treatment of the few reported cases has been variable and producing varying outcomes.
Generalized lymphatic anomaly
GLA is a multifocal lymphatic disorder frequently presenting with focal lytic lesions in multiple bones, typically the ribs and the spine, without infiltrating soft tissue mass or loss of cortical bone. Of note, current literature suggests avoidance of biopsy of rib lesions as this can lead to recurrent PLeff as seen in case 2 of this series. 13 While bony changes are well visualized on CT, MRI is required to assess bone marrow and soft tissue involvement. Medical therapy with interferon and bisphosphonates, and in recent years, sirolimus has provided great relief to patients. 14
Kaposiform lymphangiomatosis
KLA is a unique disease with clinical and imaging findings commonly overlapping with GLA. In KLA, perivascular congested soft tissue is noted on MRI, often with significant thoracic involvement that enhances with contrast. 15 The associated lytic osseous lesions are cortex-sparing and noncontiguous, and other common findings include chylothorax and splenic cysts. Upon biopsy, KLA shows malformed lymph vessels with diagnostic spindle-shaped lymphatic endothelium and focal areas of proliferation. 16 Patients may present with extravascular hemorrhage, hemothorax, thrombocytopenia, and coagulopathy in addition to localized pain over the locations of bony involvement. Although an aggressive disease, sirolimus has helped achieve stable disease status in some cases. 17
Diagnostic dilemma
Perhaps, the one common theme among patients in our small series and the experience of others is the diagnostic challenge for cases involving CLA. Patients can experience a variety of symptoms and are initially evaluated by different medical specialties, and the lack of a uniform approach to diagnose CLA often leads to delayed treatment. Iacobas et al. describe guidelines that incorporate a multidisciplinary approach to the initial evaluation of suspected CLA. 18 With continued improvement in imaging modalities, a combination of radiography, pathology, coagulation profile, and clinical features can help aid in proper diagnosis.
Imaging pattern has been especially useful in distinguishing GLA and GSD, with the latter being associated with progressive osteolysis with loss of cortical bone. In GLA, the osseous involvement tends to be more generalized with dilated lymph channels and preservation of the cortex with a pattern of bony involvement that may be noncontiguous with frequent involvement of the appendicular skeleton in the form of “punched out” lesions.13,15
Although there is considerable imaging overlap between KLA and GLA, the former is typically characterized by extensive thoracic involvement along with elevated D-dimer, prolongation of prothrombin time or partial thromboplastin time, and low fibrinogen with thrombocytopenia.
Case 1 was initially diagnosed with GSD based on cortical bone involvement of the base of the skull seen both on imaging and biopsy as well as the absence of laboratory abnormalities typically seen in KLA. However, <1 year later, she presented again with a soft tissue mass in the neck as well as coagulopathy and thrombocytopenia, both of which were concerning for KLA. A biopsy of the neck area showed only distended lymphatics without the foci of dense spindle-cell D2-40 positive vascular channels that are diagnostic for KLA. However, because of her clinical features and laboratory abnormalities, her treatment was guided toward KLA with good therapeutic response.
Case 2 initially presented with generalized symptoms including back pain and shortness of breath. After undergoing imaging and biopsy, she was thought to have GSD due to the diffuse pattern of bone loss replaced by lymphatic channels throughout her thoracic spine. However, upon further analysis, we feel that her diagnosis was more reflective of GLA due to the large recurrent PLeffs and the fact that her bone lesions appeared to be multiple and discrete rather than one contiguous lesion as seen in GSD.
In our third case, the patient's diagnosis was likely delayed by several years; unfortunately, he underwent several surgical procedures before ultimately being diagnosed with GSD. This was then followed by a period of several years until GSD specific therapy could be prescribed.
Medical treatment options
Sirolimus
As an inhibitor of mTOR—a kinase involved in the PI3K/Akt pathway—sirolimus has been shown to inhibit multiple cellular processes including angiogenesis and cell growth. Several articles have demonstrated the efficacy of sirolimus in treating lymphatic abnormalities including Hammill et al. who successfully treated four patients with microcystic lymphatic malformations (LMs). 19 Similarly, Ricci et al. demonstrated efficacy of sirolimus in stabilizing/reducing signs and symptoms in patients with complicated lymphatic malformations. 14 In our series, all three patients were treated with sirolimus with variable response. Patients were monitored regularly for the known toxicities of sirolimus. We observed hypertriglyceridemia and mucositis, both of which were managed without altering the sirolimus course. Sirolimus goal troughs were 8–12 ng/mL for our patients who received it twice daily.
Bisphosphonates
In CLA—and GSD in particular—there is massive osteolysis secondary to abnormal proliferation of blood or lymphatic vessels leading to replacement of bone marrow spaces. 9 This process is possibly driven by high levels of interleukin-6, which triggers formation and activation of osteoclasts. 10 Thus, bisphosphonates have been used in this setting due to their anti-osteoclastic properties. Newer generation bisphosphonates also have anti-VEGF effects that may be helpful in CLA management. 9 In fact, our patients achieved significant relief of symptoms from the use of bisphosphonates in conjunction with other agents. This experience is similar to that of others who have successfully used bisphosphonates in patients with GSD.9,20 Patients receiving bisphosphonate therapy need to be monitored for known side effects, and dental examination should be performed for risk of osteonecrosis. For our patients, normal serum calcium and phosphorus were noted throughout treatment without any sign of intolerance or toxicity. For case 1, transient elevation of intact parathyroid hormone was noted. A known risk of treatment-related fractures and jaw osteonecrosis exists with ZA use. Fortunately, none of these side effects have been observed among our patients despite prolonged use of ZA in the first two cases. After discussing the relevant risks versus benefits with the patients and their families, it was decided that the benefits of continued ZA treatment far outweighed the risks.
All three of our patients appeared to benefit greatly from the addition of bisphosphonates despite their various diagnoses. Case 1 initially received IV pamidronate. Upon progression, she was immediately started on IV ZA. Fortunately, the next set of images showed that her bony disease had stabilized, and now she is successfully maintained on an annual dosing regimen of ZA.
Case 2 was started on monthly IV ZA at the time of diagnosis due to the severity of her bony disease. After the first 3 months, imaging showed stabilization of her bony disease allowing discontinuation of ZA. Two years later, she presented with relapse of her symptoms, and again, she resumed once monthly ZA. Once her disease stabilized, doses were gradually spaced out.
Patient 3 required only three doses of once daily IV pamidronate to achieve stabilization of disease. This may be due to the fact that he underwent a concurrent amputation of the affected bone.
Vincristine
Patients with GLA have been historically managed with supportive care and interventions guided by anatomical involvement and symptoms. The vinca alkaloid vincristine is widely used by oncologists to treat a variety of malignancies; it is an antiangiogenic agent that acts via inhibition of cell mitosis and microtubule formation. 21 In our study, two of the three patients received multiple doses of vincristine at 1.5 mg/m2 with good tolerance. Specifically, earlier in treatment, patient 1 received weekly doses of vincristine for 6 months with multiple unsuccessful attempts at spacing out doses. Fortunately, she is now well controlled without the use of vincristine. Case 2 required weekly vincristine for 6 months without recurrence of symptoms following completion of treatment. Common adverse effects such as peripheral neuropathy, jaw pain, or electrolyte imbalance were not noted.
We note that response to vincristine can be variable: Ludwig et al. described a case of GLA that did not respond well. 22 However, there is literature supporting the use of sirolimus, often with steroids, with or without vincristine to treat KLA. 23
Steroids
Steroids as well as other agents such as bisphosphonates, calcitonin, and vitamin D have been described to treat patients with CLA and osteolysis. 24 Additionally, steroids have been used to treat microcystic LMs as these are more infiltrative, making surgical resection less feasible. 25 For patients with kaposiform hemangioendothelioma with Kasabach–Merritt syndrome, a multidisciplinary consensus statement recommended use of steroid and vincristine as first-line therapy. 23 In our series, case 1 has required a multitude of medical interventions including the use of steroid with/without vincristine to manage flares of KLA. The medication was well tolerated without severe side effects.
Multiagent therapy
Another clinical challenge when managing patients with CLA is lack of consensus guidelines with regard to therapy. Treatment of CLA is often guided by anatomical location and degree of involvement. Procedures such as drainage of PLeff as in case 2 are mainly aimed at providing symptomatic relief, although recurrence may occur. Medical management can vary widely and requires keen attention and ability of the clinical team to adjust and reinforce treatment when necessary, exemplified among the first two patients in our series who required substitution of agents as well as reinforcement when appropriate. In the end, none of the patients could receive “standard” or “uniform” treatment but achieved signs of improved quality of life with less pain and fewer hospitalizations. The patients in our series were treated with a combination of bisphosphonate and sirolimus therapy. In fact, patients 1 and 2 responded well to treatment with ZA. The use of bisphosphonates is a less reported aspect of therapy for patients with CLA. Our small series shows it to be both efficacious and well tolerated.
With regard to supportive care, all three patients were treated with Bactrim for Pneumocystis carinii prophylaxis, which was well tolerated.
Limitations
Our study is limited by small sample size, perhaps related to the rarity of CLAs. We look forward to further studies (both clinical trials and basic science) that need to be performed to determine the optimal diagnostic and therapeutic strategies for this devastating disease. Also, because our study was a retrospective chart review, we did not have definitive time points by which to measure the response of disease to therapy or have predefined definitions to assess quality-of-life measurements for all of our patients.
Conclusion
CLA encompasses a unique variety of clinical scenarios with overlapping features, making diagnosis and treatment difficult for each individual case. This case series of three patients depicts a multidisciplinary and multiagent approach to the management of CLA with severe bony involvement using sirolimus, bisphosphonates, vincristine, and steroids.
Footnotes
Authors' Contributions
Z.M.J. and A.R. initiated the project design. M.D.A., A.S., and Z.M.J. collected data and literature review for the study and drafted the article. L.R.M. contributed the pathology images and diagnoses. S.F. contributed the radiology images. K.H., L.J.A., and A.R. provided clinical history of patients treated. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this study.