
Abstract
CCL22 inactivation in vivo occurs by cleavage at the N-terminus; however, it is unclear whether this encompasses the entire site of CCR4 interaction. CCL17 also binds CCR4 and its function requires binding via two discrete binding sites. Using monoclonal antibodies (MAbs), we report that there are two separate sites on CCL22 that are required for CCR4-mediated function. The CCL22-specific antibodies bind with affinities of 632 ± 297 pM (MC2B7) and 308 ± 43 pM (MAB4391) and neither exhibited detectable binding to CCL17. Both antibodies are comparable in their ability to inhibit CCL22-mediated calcium mobilization; however, competition binding studies demonstrate that MC2B7 and MAB4391 bind to distinct epitopes on CCL22. Both antibodies inhibit function through CCR4, which is demonstrated by loss of β-arrestin recruitment in a reporter cell line. In both assays, blocking either site independently abolished CCL22 function, suggesting that concurrent engagement of both sites with CCR4 is necessary for function. This is the first demonstration that CCL22 has two distinct binding sites that are required for CCR4 function. These antibodies are valuable tools for better understanding the interaction and function of CCL22 and CCR4 and will potentially help further understanding of the differential outcomes of CCL17 and CCL22 interaction with CCR4.
Introduction
A
In contrast to CCL17, which is primarily associated with maintaining localized inflammation, CCL22 has emerged as a down modulator of inflammation. Regulatory T cells (TREG) present in ovarian tumors are associated with CCL22, which is produced by tumor-associated macrophages and DCs.(16) CCL17 was also detectable but produced at lower levels in these tumors, implicating a role for CCL22 in maintaining a TREG population. Analysis of human breast tumors also shows a correlation between CCL22 and CD25+ CD4+ Foxp3+ T cell co-localization.(17) In lung cancer patients, CCL22 is predominantly elevated in pleural fluid compared to levels in sera, and purified TREG cells are differentially inhibited by anti-CCL22 rather than anti-CCL17 ex vivo.(18) A role for CCL22 in down-modulating the immune response through TREG cells has also been demonstrated in the context of allograft tolerance. Expression of CCR4 is required for maintenance of tolerance in a mouse cardiac allograft model, and CCL22 expression is preferentially increased in conditions of long-term tolerance induction whereas CCL17 is not.(19) This is supported by the finding that expression of indoleamine 2,3-dioxygenase (IDO) correlates with CCL22 in DCs isolated from gut-associated lymph nodes and CCL22 is induced upon exposure of DCs to apoptotic cells.(20) In the NOD mouse model of diabetes, enhancement of CCL22 expression through introduction of a transgene increases the TREG population in the pancreas, which subsequently prevents development of diabetes.(21) This is somewhat paradoxical since others have reported that introducing a CCL22 transgene into mice from birth accelerates the onset of diabetes by increasing the influx of T cells into the pancreas.(22) The role of CCL17 in these models has not been reported.
It is becoming clear that CCL22 is an important regulator of the innate immune response.(3,23,24) In a study of the murine cecal ligation and puncture (CLP) model of experimental sepsis, the investigators demonstrated that administration of exogenous CCL22 was able to protect mice from CLP-induced mortality through enhancement of bacterial phagocytic and killing activity of macrophages. Neutralization of CCL22 in this model resulted in reduced recruitment of macrophages and increased bacterial load.(23) While the role of CCL17 was not explored in this system, blockade of CCL17 has been shown to be protective in a model of invasive Aspergillosis, while neutralization of CCL22 in the same model inhibited anti-fungal responses.(25) More recently, using the A. fumigatus model of allergic airway inflammation, a commercially available monoclonal antibody specific for murine CCL22 failed to inhibit airway hyper-responsiveness (AHR).(26) This is in contrast to what was observed following neutralization of CCL17 in the same model system.(26,27)
Following four rounds of phage library panning, a panel of eight CCL22 specific antibodies was identified from which three unique clones were chosen for in vitro characterization. Based on the ability to block CCL22-mediated calcium flux and β-arrestin recruitment comparably to a commercially available antibody, MAB4391, we characterized the CCL22 binding characteristics of two of these antibodies and established that MC2B7 recognizes a distinct epitope from MAB4391. We demonstrate herein that inhibition of CCL22 function is mediated by blocking either site.
Materials and Methods
Generation of anti-mouse CCL22 antibodies
Bt-CCL22 (100 nM for rounds 1 and 2, 10 nM for rounds 3 and 4) was captured on streptavidin (SA)-coated magnetic beads (Dynal, M280), washed with 3x with PBST (PBS with 0.05% Tween-20), and blocked for 1 h with 1 mL PBST-M (PBST with 3% non-fat dry milk). De novo pIX antibody libraries(28) were blocked with PBST-M added to the beads and rotated at room temperature for round 1. Beads were washed three times with PBST and three times with PBS. Beads were then incubated in a culture of log phase Escherichia coli (MC1061F’) cells and the infected E. coli were grown in LB (Carb) media at 37°C until OD of 0.6. Helper phage was added to the cultures, followed by incubation at 37°C for 30 min. Kanamycin and IPTG were added to each culture to final concentrations of 35 μg/mL and 1 mM, respectively, then grown overnight at 30°C with shaking. The amplified phage from the bacterial media was precipitated using PEG/NaCl and re-suspended in 1 mL PBS. 100 μL of the phage were used for the next round of panning.
After four rounds of panning, phagemid DNA was isolated from the infected MC1061F’ cells and digested with restriction enzymes NheI and SpeI to remove the sequence encoding pIX. The restriction digestion was run on a 1% agarose gel and the linearized plasmid DNA was excised and purified using Qiaquick gel extraction columns. This DNA was then self-ligated with T4 DNA ligase, resulting in an in-frame His-tag at the C-terminus of the Hc. The ligated DNA was electroporated into MC1061F’ cells and plated onto LB (Carb/glucose) agar plates.
Fab expression and antigen binding ELISAs were performed. Colonies were picked and grown in 1000 μL 2xYT/Carb at 37°C during the day until their OD600nm reached approximately 1.0. IPTG was added to a final concentration of 1 mM, and cultures were grown overnight at 30°C with shaking. Maxisorp 96-well plates were coated with either 5 μg/mL sheep anti-human-Fd (Binding Site, San Diego, CA) or 5 μg/mL of streptavidin. To the streptavidin-coated plates, 2 μg/mL Bt-CCL22 were added to display the antigen for Fab binding. Plates were blocked in PBST-M. Cultures were centrifuged at 3500 rpm for 10 min. Spent culture supernatant containing Fab was added to both the bt-huCCL17 displayed plate and the anti-human Fd-coated plate. Anti-human Kappa-HRP (1:5000 in PBST; Southern Biotech, Birmingham, AL) was added to detect Fabs in both ELISAs. Finally, the chemiluminescence substrate PoD (Roche, Indianapolis, IN) was added, and the plates assayed in an EnVision plate reader (Perkin Elmer, Waltham, MA).
Calcium flux assays
Calcium flux mobilization assays using CCRF-CEM (ATCC# CCL-119) were performed as previously described.(27) Briefly, cells were cultured in RPMI with GlutaMAX, 10% FBS, 10 mM Hepes, 1 mM sodium pyruvate, 4500 mg/L glucose, and 1500 mg/mL sodium bicarbonate at 37°C incubator with 5% CO2. The cells were harvested by centrifugation, resuspended at 1.6 × 106 cells/mL, and then seeded in 384-well poly-D-lysine–coated black view assay plate (Greiner Bio-One, Monroe, NC) in the presence of Fluo-8 NW dye-loading buffer (ABD Bioquest, Sunnyvale, CA); the buffer consisted of 10 μL Fluo-8 NW, 1 mL 10x Pluronic F127 Plus, and 19 mL assay buffer with 0.1% BSA, which was allowed to incubate at 37°C with 5% CO2 for 30 min. Purified phage α-CCL22 MAbs, MAB4391 control MAb (R&D Systems, Minneapolis, MN), rat IgG2A isotype control MAB006 (R&D Systems), or assay buffer was pre-incubated with mCCL22 for 60 min at room temperature. Calcium mobilization was assessed using FDSS 6000 (Hamamatsu, Bridgewater, NJ) to measure the fluorescence intensity using 490 nm excitation and 525 nm emission.
β-arrestin
The β-arrestin recruitment assay was performed as described previously using human CCR4 expressing CHO cells according to the manufacturer's recommendations (PathHunter eXpress β-arrestin Human and Ortholog GPCR assay, DiscoveRx Corp., Freemont, CA).(29) Mouse CCL22 was pre-incubated in the presence or absence of the antibodies from the anti-CCL22 panel, control anti-CCL22 MAB4391 (R&D Systems, Minneapolis, MN), or isotype control antibodies rat IgG2a, CNTO1488, or control rat-mouse chimeric IgG1/κ (CNTO5516) for 20–30 min at 37°C with 5% CO2 before addition to cells. Following addition, the assay was allowed to proceed for 90 min. Chemiluminescent detection substrates were added and incubated at room temperature for 60 min. Luminescence readings were taken on the Victor3 V Wallac1420 multi-label counter (Perkin Elmer, Santa Clara, CA) and analyzed on GraphPad Prizm software. For dose curves, concentration started at 1 μM with a 1:2 dilution series. For antibody inhibition studies, mouse CCL22 was used at a constant concentration of 20 nM.
Binding affinity
The binding affinities were determined either by kinetic experiments using SPR or by solution equilibrium affinity (SEA). SPR experiments were performed using a ProteOn SPRX36 optical biosensor (Bio-Rad, Hercules, CA). A ProteOn biosensor surface was prepared by coupling approximately 2500–2700 RU of goat anti-rat Fc antibody (cat. # 112-005-071, Jackson ImmunoResearch, West Grove, PA) onto a ProteOn GLC chip by standard amine coupling chemistry according to manufacturer's instructions. Kinetic experiments were run in running buffer (PBS [pH 7.4], supplemented with 0.05% surfactant P20, 200 μg/mL BSA, and 3 mM EDTA) at 25°C. The anti-CCL22 MAbs were captured (100–150 RU) onto the α-rat Fc antibody-modified sensor chip surface. Capture of anti-CCL22 MAbs was followed by injection of murine CCL22 in solution (500 nM, 100 nM, 25 nM, 6.3 nM, 1.6 nM, and 0 nM). Antigen association was monitored for 5 min at a flow rate of 60 μL/min, and dissociation was monitored for 1 h. The chip surface was then regenerated to immobilized goat anti-rat Fc using short pulses of 100 mM H3PO4. Data were double referenced to chip interspots and to buffer response to correct for buffer contribution to the signal and instrument noise.(30) Data were fitted using a Langmuir 1:1 model with kinetic constants and Rmax grouped, unless otherwise stated.
For solution equilibrium affinity (SEA) measurements, the Meso Scale Discovery (MSD, Gaithersburg, MD) reader was used for detection (MSD-SEA); 6-fold serial dilutions of CCL22 at a starting concentration of 2 mM were prepared in Tris-based saline buffer containing 0.05% Tween-20, TBST (Thermo Scientific, Somerset, NJ). Equal volumes of each MAb at 200 pM were added to each dilution in duplicate, then incubated at 4°C for 48 h. MSD-Streptavidin High Bind MA6000 96-well plates (MSD) were blocked with TBST for 2 h at room temperature, then washed three times with MSD wash buffer. Biotinylated mouse CCL22 was then added to the MSD-SA plates at 25 ng/mL and incubated at room temperature for 1 h, then washed three times before addition of the mCCL22/MAb mixtures followed by incubation for 1 h. Plates were then washed three times and ruthenylated F(ab’)2 donkey α-mouse IgG (H+L) was added followed by incubation for an additional 30 min with moderate shaking at room temperature. After washing three times, MSD Read Buffer T with surfactant, prepared according to manufacturer's directions, was dispensed to each well and analyzed with a Sector Imager 6000 (MSD).
Competition binding studies
ELISA binding of mouse CCL22 with MAbs
High bind plates (Meso Scale Discovery, Gaithersburg, MD) were coated directly with 10 μg/mL anti-mouse CCL22 antibodies MC2B5, MC2B7, MAB4391, and CNTO607 (negative control in 0.1 M HEPES buffer [pH 7.4]), then blocked with 5% MSD Blocker A buffer for 2 h at room temperature. After three washes with 0.1 M HEPES buffer (pH 7.4), MSD Sulfo-tag labeled mouse CCL22 was serially diluted 2-fold (from 200 nM to 100 pM), added to the designated wells, and incubated for 2 h with shaking at room temperature. After the incubation, plates were washed three times with 0.1 M HEPES buffer (pH 7.4). MSD Read Buffer T was diluted with distilled water (4-fold) and dispensed to each well, then analyzed with a MSD Sector Imager 6000.
Competition ELISA
Competition binding assays were performed to evaluate differential binding epitopes of anti-mouse CCL22 antibodies. Recombinant mouse CCL22 (10 μg/mL) was directly coated on MSD high bind plates for 2 h at room temperature, then blocked with 5% MSD Blocker A buffer for an additional 2 h at room temperature. Plates were washed three times with 0.1 M HEPES buffer (pH 7.4), followed by addition of the mixture of 10 nM labeled MC2B7 antibody with different competitors. Labeled antibody was incubated with increasing concentrations of competitors, from 1 nM to 1 μM, and then added to the designated wells. After incubation with gentle shaking at room temperature for 2 h, plates were washed three times with 0.1 M HEPES buffer (pH 7.4). MSD Read Buffer T was diluted with distilled water (4-fold) and dispensed into each well and analyzed with a MSD Sector Imager 6000.
Alternatively, anti-mouse CCL22 antibody MAB4391 (10 μg/mL) was directly coated on MSD high bind plates for 2 h at room temperature, then blocked with 5% MSD Blocker A buffer for an additional 2 h at room temperature. Plates were washed thee times with 0.1 M HEPES buffer (pH 7.4), followed by addition of the mixture of 40 nM labeled mouse CCL22 with different antibodies as competitors. Labeled mCCL22 was incubated with increasing concentrations of competitors, from 1 nM to 1 μM, and then added to the designated wells. After incubation with gentle shaking at room temperature for 2 h, plates were washed three times with 0.1 M HEPES buffer (pH 7.4). MSD Read Buffer T was diluted with distilled water (4-fold) and dispensed into each well and analyzed with a MSD Sector Imager 6000.
Results
CCL22 function is inhibited in vitro by chimeric MAbs having mouse IgG1 isotype
In order to create an improved anti-CCL22 antibody, de novo panning using a human Fab phage library was used to generate monoclonal antibodies. A panel of eight CCL22-specific antibodies was identified and subsequently characterized for ability to inhibit CCL22-mediated calcium mobilization. A dose response curve was run for each antibody in the panel and the results showed that five of the eight antibodies inhibited CCL22 function in this assay: MC2B3, MC2B4, MC2B5, MC2B6, and MC2B7 (Fig. 1). No inhibition was seen with MC2B1, MC2B2, or MC2B8. The dose response curves generated for the neutralizing antibodies are distinct but overall potency does not appear to vary much between antibodies in this assay. Differences become more evident when comparing the IC50 of each antibody (Table 1), which reveals an approximately three-fold difference between the highest and lowest values although all IC50 values are superior to that of MAB4391, which is 82.99 ng/mL. Upon sequence analysis of VH and VL chain genes used by each of the neutralizing chimeric antibodies, it was determined that three different VL genes were represented among the five clones. For this reason, only the three unique clones, MC2M3, MC2M5, and MC2M7, were further characterized.

Anti-CCL22 specific antibodies inhibit calcium mobilization in a dose-dependent manner. CCRF-CEM cells were treated with recombinant mouse CCL22 at 3 ng/mL as described in Materials and Methods section. Antibodies are as follows: MC2B1 (solid triangles), MC2B2 (solid triangles, inverted), M2B3 (solid diamonds), MC2B4 (solid circles), MC2B5 (solid squares), MC2B6 (open triangles), MC2B7 (open triangles, inverted), MC2B8 (open diamonds). Controls include cells incubated in the absence of CCL22 (×), indicating baseline calcium flux, and cells incubated with CCL22 in absence of antibody (star), indicating maximum calcium flux.
The only known receptor for CCL22 and CCL17 is CCR4, which is a Gi coupled G protein-coupled receptor (GPCR) that recruits β-arrestin immediately upon interaction with either chemokine.(1,31–33) To verify that the functional inhibition afforded by these MAbs is due to specific interference with CCL22 signaling through CCR4, β-arrestin recruitment was monitored using a reporter cell line, as described for CCL17.(29) This cell line expresses the human CCR4 receptor and produces a signal that is proportional to the amount of β-arrestin recruited upon engagement. Antibodies were tested in dose response curves at concentrations ranging from 1 nM to 1 μM using a constant CCL22 concentration of 20 nM. Inhibition of β-arrestin recruitment was seen with all three antibodies and similar to results from the calcium mobilization assay, MC2B3 appeared slightly less effective than the MC2B5 and MC2B7 (Fig. 2). MAB4391 was also tested in parallel and exhibits inhibitory activity comparable to MC2B5 and MC2B7. Because this reporter cell line is designed such that a signal is only generated through CCR4 engagement, these data indicate that the antibodies being tested are blocking CCL22 function by interfering with the ability to signal through CCR4.

Anti-CCL22 antibodies inhibit CCR4-mediated function of CCL22. Recruitment of β-arrestin in response to CCL22 is inhibited in a dose-dependent manner by IgG1 chimeric and rat IgG2a α-CCL22 specific MAbs. Antibodies were incubated with a human CCR4 reporter cell line in the presence of 20 nM recombinant CCL22. Shown are data for MC2B3 (circles), MC2B5 (solid triangles), MC2B7 (squares), and MAB4391 (open triangles). Controls include cells incubated in the absence of CCL22 (×), indicating baseline signal, and cells incubated with CCL22 in the absence of antibody (star), indicating maximum β-arrestin recruitment. Antibodies were tested in this assay twice and data shown represent one of the experiments.
MAB4391 and MC2B7 possess different CCL22 binding profiles even though they both inhibit CCL22 function in vitro
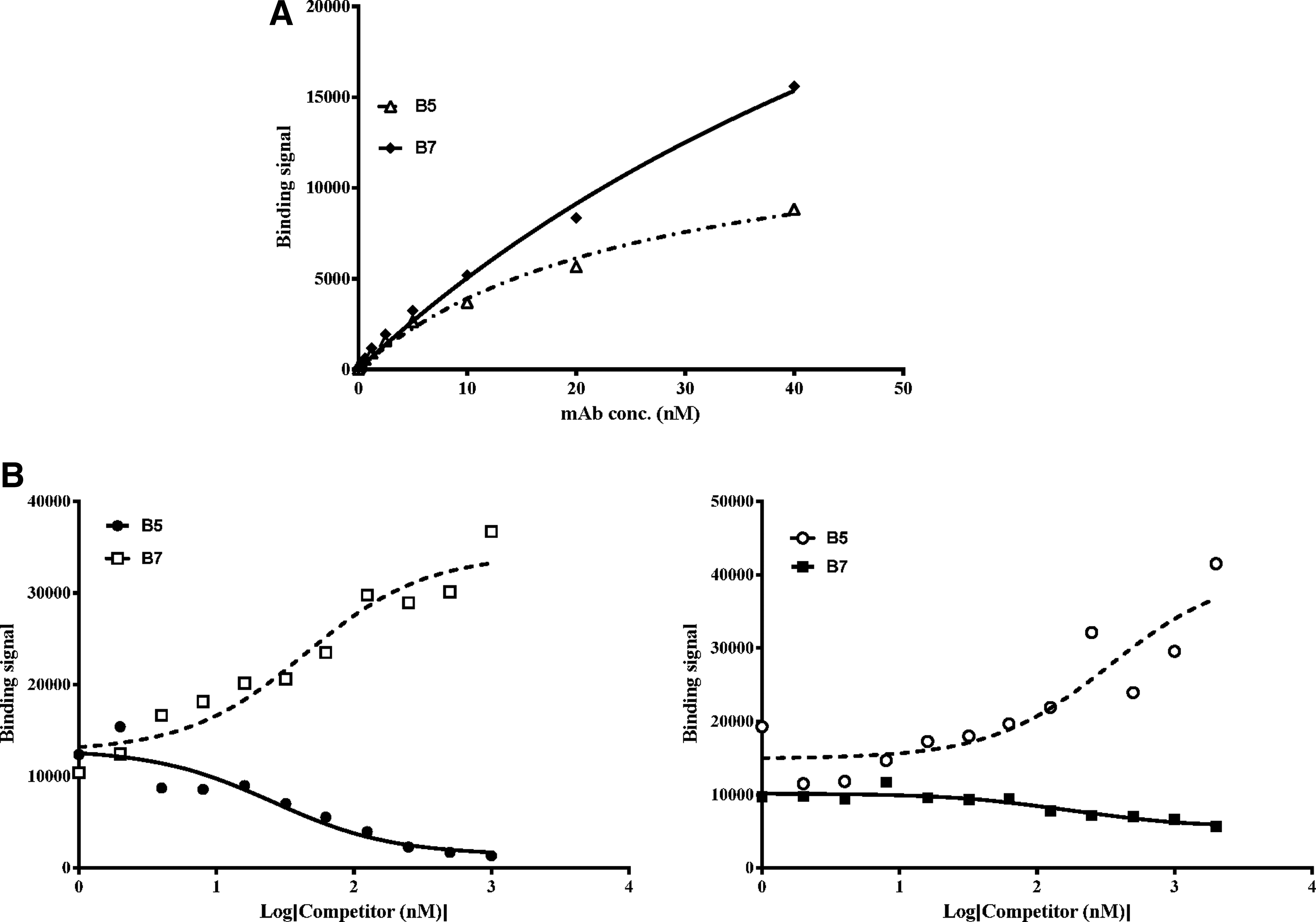
Epitope and binding affinity are contributing factors in determining how well the antibodies inhibit CCL22 function. Previously, we identified two antibodies that effectively inhibit CCL17 but each antibody recognizes a distinct epitope.(27) Through binding studies, we were able to show that the antibodies bind to non-overlapping epitopes on CCL17 with KDs differing by greater than 7-fold. To determine whether the anti-CCL22 antibodies could be similarly differentiated, ELISA-based competition assays were performed. The chimeric antibodies MC2B5 and MC2B7, as well as MAB4391, are able to bind CCL22 directly coated onto an ELISA plate; however, the binding curves are not superimposed (Fig. 3A). Differences in binding affinity could account for the binding curve incongruence but the affinities of these three antibodies for CCL22 are within 3-fold, suggesting there is another factor at play (Table 2). A likely explanation for the differences is that each antibody recognizes a different epitope and that the MC2B5-specific epitope is fully intact whereas those recognized by MC2B7 and MAB4391 are perturbed when CCL22 is plate-bound. Competition binding studies in which MAB4391 is used to compete with labeled MC2B7 (Fig. 3B) clearly demonstrate that MAB4391 and MC2B7 do not compete for binding to plate-bound CCL22. The fact that these two antibodies recognize distinct epitopes is established in Figure 3C, which shows that CCL22 captured by plate-bound MAB4391 binds MC2B7 whereas binding by MC2B5 is hindered. Analogous to MAB4391, MC2B5 fails to compete with MC2B7 for CCL22 binding (Fig. 4), though all three antibodies inhibit CCL22 function.
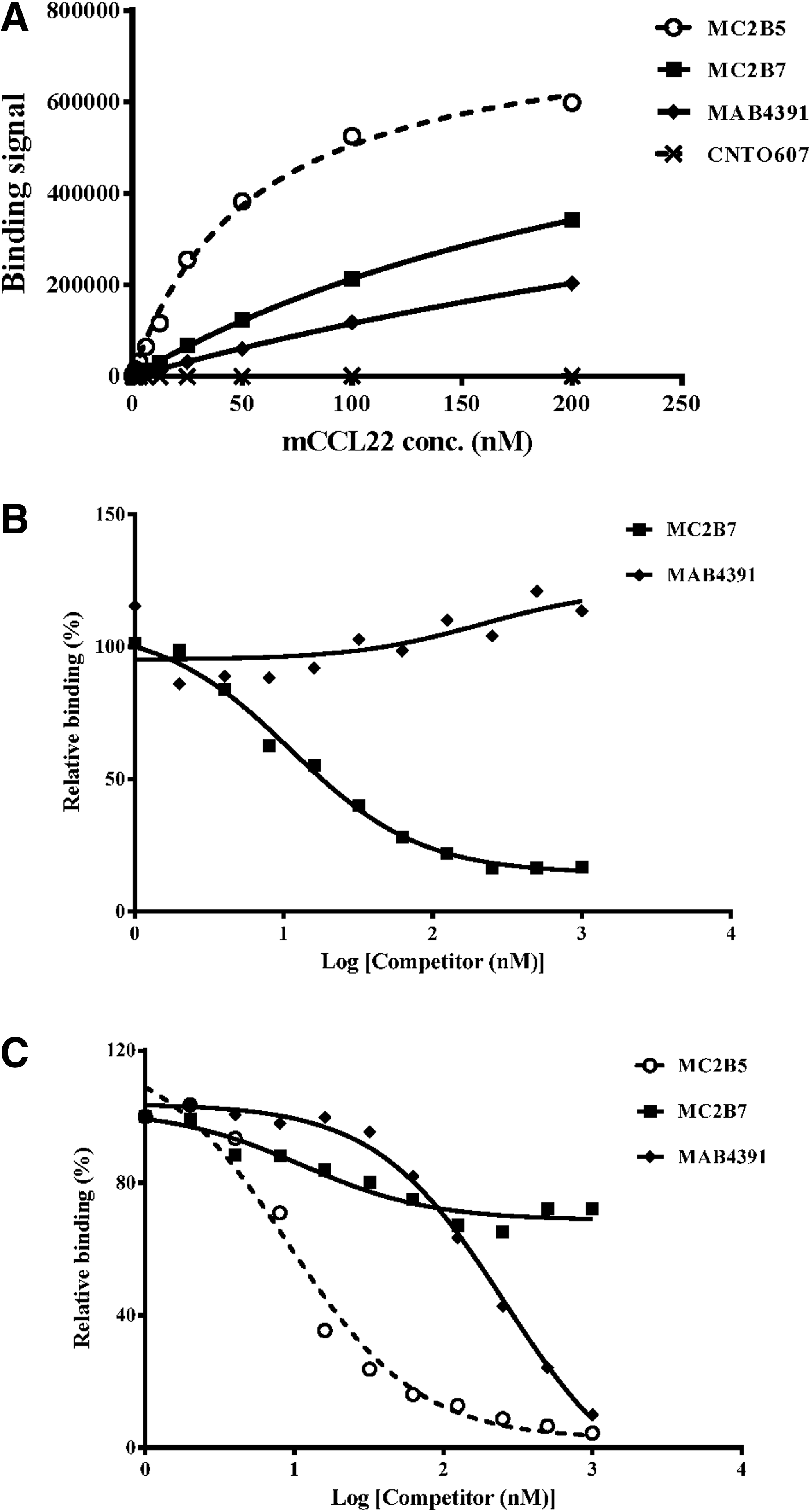
Each antibody binds a unique and non-overlapping epitope on CCL22. All three antibodies bind to plate-bound CCL22, as measured using ELISA format (
MC2B5 and MC2B7 inhibit CCL22 function through interaction with distinct epitopes. (
ND, not determined.
Discussion
Functional activity of CCL22 can be inhibited using MAbs that recognize either one of two non-overlapping sites; also blocking either site impairs function. The previously reported lack of in vivo impact with anti-CCL22 treatment using MAB4391(26) raised questions as to whether MAB4391 has potent inhibitory activity. Two parallel strategies were pursued to address this question: generation of additional anti-CCL22 antibodies and confirmation of the inhibitory activity of MAB4391 in vitro. A set of three chimeric antibodies, MC2B3, MC2B5, and MC2B7, was created and shown to effectively neutralize functional activity of mouse CCL22 in vitro using two different assays—calcium mobilization and β-arrestin recruitment. MAB4391 was used as a comparator for our experiments and demonstrated similar efficacy in each assay. Of the chimeric antibodies, MC2B7 had properties most closely resembling MAB4391, with the exception of having better inhibitory activity in the calcium flux assay.
Competition binding studies reveal that although MC2B7 performs comparably to MAB4391 in vitro, this antibody binds CCL22 via a distinct epitope, suggesting that CCL22 has two separate binding sites for CCR4 and both must be engaged in order to mediate function. Using monoclonal antibodies, we previously demonstrated that CCL17 requires interaction with at least two sites for CCR4-mediated signaling to occur.(27) CCL22 and CCL17 have been reported to compete with each other for CCR4 binding, suggesting that these ligands interact with overlapping sites on the receptor.(1,7,32,34) Yet when generating the anti-CCL17 antibody panel, we were only able to identify a single antibody in our screen that exhibited cross-reactivity with both.(29) This suggests that CCL17 and CCL22 may interact with CCR4 through a common site but also interact with a second site that is unique to each chemokine, ultimately owing to the differences in function.
CCL22 is more tightly regulated than CCL17 in vivo, suggesting a key role for CCL22 in immune modulation. Cleavage of the N-terminal amino acids 1–4 by CD26 renders CCL22 inactive as this ablates chemotactic activity and calcium mobilization in CCR4+ cells.(35) In addition to cleavage, sequestration is also a mechanism for regulating chemokine function. The chemokine scavenging receptor D6 preferentially binds CCL22 although it regulates activity of both CCL17 and CCL22. However, only full length CCL22 is recognized by D6, thus providing an additional mechanism for controlling activity.(36) Although CCL17 is also subject to control via sequestration through interaction with the Duffy antigen receptor for chemokines (DARC), this receptor does not bind CCL22.(37) That there are several mechanisms in place to prevent accumulation of active CCL22 suggests an acute sensitivity to functional activity so that any elevation in levels of this chemokine is detrimental. Perhaps a key role of CCL22 in mediating innate immunity is dependent on a minimal level of active chemokine such that complete inhibition (e.g., by MAB4391 or MC2B7) is also detrimental.
Additional characterization of the structural properties of MC2B7 and MAB4391 to better understand interaction with CCL22 could also help in understanding the functional properties. Although they bind distinct epitopes on CCL22, it is unknown whether either of these antibodies binds the N-terminus of the full length chemokine. Both inhibit CCL22 function in vitro and the prediction is that one of these antibodies interacts with the N-terminus. Adding to the complexity, CCL22 functional activity has been reported to increase upon interaction with CXCL10, which is mediated via CCR4.(38) Although the amino acids involved in the interaction with CXCL10 have not been determined, it would be interesting to see if this interaction was maintained in the presence of either MC2B7 or MAB4391 as well as cleaved CCL22.
Footnotes
Acknowledgments
We wish to thank Bingyuan Wu for his help generating recombinant CCL22. We are also grateful to Les Gutshall for help with the antibody expression work.
Author Disclosure Statement
The authors have no financial conflicts to disclose.