
Abstract
SNAP-tag technology allows recombinant proteins to be covalently labeled to O6-benzylguanine (BG)-modified substrates with 1:1 stoichiometry. By attaching according fluorophores, this method is ideally suited for in vitro and in vivo imaging, as well as protein interaction analyses. Fluorophores modified with BG react with the SNAP-tag, whereas those modified with O2-benzylcytosine (BC) conjugate to a more recent derivative known as the CLIP-tag. The orthogonal substrate specificity of the SNAP- and CLIP-tags extends the range of applications by allowing double labeling. We previously developed a monoclonal antibody (mAb) that recognizes both tags. In this study, we describe a new mAb, which is specific for the SNAP-tag alone. Therefore, this mAb allows discrimination between SNAP- and CLIP-tags within a broad range of immunological methods, including enzyme-linked immunosorbent assays, western blotting, flow cytometry, and immunohistochemistry.
Introduction
T
SNAP-tag technology allows proteins to be labeled using dyes modified with O6-BG for in vitro or in vivo imaging, and this approach is suitable for the analysis of protein functions, protein trafficking, and protein–protein interactions.(9–15) For example, the SNAP-tag has been fused to the membrane-bound epidermal growth factor receptor (EGFR) and coupled to different BG-modified dyes, allowing the EGFR to be tracked in living cells by single-molecule fluorescence microscopy.(11) Similarly, the SNAP-tag has been fused to single-chain variable fragment (scFv) antibodies specific for prostate-specific membrane antigen (PSMA), and BG-modified dyes have been used to verify scFv internalization by PSMA-positive cells in culture.(13)
Monoclonal antibodies (MAbs) are ideal reagents for antigen-specific detection, because their homogeneity confers consistent and reproducible properties in different immunological assays.(16) Two mAbs against the SNAP-tag have been described: a single-domain (VHH) recombinant mAb generated by panning a naive Llama library(17) and mAb M2D11 generated using standard hybridoma technology.(18) The VHH mAb recognizes the SNAP-tag in immunofluorescence and immunoprecipitation procedures, but its ability to recognize the CLIP-tag and discriminate between SNAP and CLIP is unknown. In contrast, M2D11 is known to bind SNAP and CLIP and can detect both native and denatured proteins containing these tags.(18)
An antibody that distinguishes SNAP and CLIP would be valuable in double-labeling experiments. Therefore, we aligned the sequences of each tag to identify unique regions and defined a SNAP-specific peptide.(19) This was injected into mice, and standard hybridoma technology was then used to generate the novel mAb anti-SNAP-11.(20) Further analysis confirmed that anti-SNAP-11 specifically recognizes the SNAP-tag, but not the CLIP-tag, and can also distinguish between the original SNAP-tag and the SNAPf variant.
Materials and Methods
Identification of a SNAP-specific peptide for immunization
Mice were immunized with a SNAP-specific peptide sequence with 18 amino acids in length (HRVVSSSGAVGGYEGGLC) showing high diversity between SNAP and CLIP, which was identified by protein alignment (CLC Main Workbench v7.5; QIAGEN). The peptide was synthesized by GeneCust and 2 mg was coupled to keyhole limpet hemocyanin (KLH) as a carrier to increase the immune response in mice(21) using the Imject Maleimide-activated Carrier Protein Spin Kit (Pierce, Rockford, IL) according to the manufacturer's instructions. The concentration of KLH-SNAPpeptide was determined using the Pierce BCA Protein Assay Kit with KLH as a standard (Sigma-Aldrich). The KLH-SNAPpeptide conjugate (4.4 μg/μL) was stored at −20°C.
Monoclonal antibodies
We immunized a 6- to 8-week-old female BALB/c mouse with 60 μg of the KLH-SNAPpeptide mixed with GERBU MM adjuvant (GERBU Biotechnik GmbH) according to the manufacturer's recommendations, followed by 30 μg boosts. The antibody titer was determined by enzyme-linked immunosorbent assay (ELISA) after high binding plates (Greiner Bio-One) were coated with 50 ng/well of the coupled or uncoupled peptide. The immunized mouse was sacrificed when the serum titer reached the range 1:51,200–1:102,400. To generate hybridoma cell lines producing an anti-SNAP mAb, the mouse spleen and the immortal mouse cell line Sp2/0-Ag14 (ATCC: CRL-1581) were prepared as previously described.(20) The mAb was purified from cells cultivated in the ISF medium (Biochrom) by Protein A chromatography, and the concentration was determined using the Pierce BCA assay.
Characterization of V-genes
Total hybridoma cell RNA was isolated (NucleoSpin® RNA II, Macherey-Nagel) and transcribed into cDNA (SuperScript™ III CellsDirect cDNA Synthesis System; Invitrogen). The V-genes were identified by polymerase chain reaction using different primer mixes,(22) followed by sequencing on an ABI PRISM® 3730 Genetic Analyzer (Fraunhofer IME).
Expression and purification of reference proteins
Six reference proteins were expressed in HEK293T cells for the characterization of the novel antibody: scFv425-SNAP, scFv425-CLIP, SNAP-EGF, CLIP-EGF, SNAPf-scFv425, and anti-HER2scFv-SNAP. Five proteins bind to the EGFR, scFv425 as a recombinant antibody fragment(23) and EGF as the natural ligand. ScFvHER2-SNAP binds to HER2. The six genes were individually transferred to the in-house pMS vector system, which provides in-frame cMyc- and His-tags for purification and detection.(24) HEK293T cells were transfected with 2 μg DNA using 4 μL Roti-Fect® according to the manufacturer's instructions (Carl Roth). The co-expression of enhanced green fluorescent protein was used to determine the success of transfection and was monitored by fluorescence microscopy. The proteins were purified from the culture supernatant by immobilized metal-ion affinity chromatography using fast protein liquid chromatography (ÄKTA; GE Healthcare) or batch purification. Purity was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) as described elsewhere(25) and western blotting to a nitrocellulose membrane (Whatman, Schleicher & Schuell) at 350 mA for 45 minutes.(18) After dialysis against 1× phosphate-buffered saline (PBS), the protein concentration was determined using the Pierce BCA assay.
ELISA
High-binding microtiter plates (Greiner Bio-One) were coated with 50 ng reference protein per well in carbonate buffer (0.2 M Na2CO3/NaHCO3, pH 9.6) mixed 1:1 with 1× PBS. The plates were incubated either for 2 hours at room temperature or overnight at 4°C. After blocking with 200 μL 1% (w/v) BSA per well (1 hour, room temperature), we added 50 μL primary antibody (1 hour, room temperature), washed the plates with PBS containing 0.1% Tween-20 (PBST), added 50 μL horseradish peroxidase-conjugated goat anti-mouse IgG (GaM-PO) secondary antibody (Sigma-Aldrich) diluted 1:5000 (1 hour, room temperature), washed as above, and detected the signal with 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate] (ABTS; Roche) by measuring absorbance at 405 nm.
Flow cytometry
Antibody binding to different cell lines was measured using a FACSCalibur (Becton Dickinson) and data were processed using WinMDI v2.9 software. We incubated 3 × 105 cells with the SNAP/CLIP-tagged proteins and the anti-SNAP-11 and M2D11 mAbs (total volume 50 μL) and detected bound antibody by adding 50 μL phycoerythrin-conjugated goat anti-mouse IgG (GaM-PE) secondary antibody (Dianova). The cells were washed with 1× PBS and centrifuged at 300 × g for 5 minutes between incubation steps, and finally, were resuspended in 400 μL PBS.
Immunohistochemistry
Tissue sections were prepared from a tumor derived from HER2-positive embryonal rhabdomyosarcoma RD cells (ATCC: CCL-136) injected subcutaneously into BALB/c mice (LANUV number 8.87-50.10.37.09.212). We prepared 8-μm sections on a cryotome (Leica) and dried them on glass slides for several days. Before staining, the tissues were fixed in acetone for 10 minutes at room temperature, circled with a Delimiting Pen (Dako), and blocked for 1 hour in 1% (v/v) mouse serum. The sections were incubated overnight at 4°C with 30 ng/μL anti-HER2scFv-SNAP fusion protein, washed thrice with PBST, incubated with a PO-labeled secondary mAb (Roche) diluted 1:200, washed as above, and finally, washed for 10 minutes with Tris-buffered saline containing 0.1% (v/v) Tween-20 (TBST) before incubating with 3,3′-diaminobenzidine (DAB) for 10 minutes at room temperature in the dark. The reaction was stopped with water, and nuclei were stained with hematoxylin for 1–2 minutes.
Surface plasmon resonance spectroscopy
The binding of the antibody to its antigen was measured by surface plasmon resonance (SPR) spectroscopy using a Biacore T200 instrument (Biacore; GE Healthcare). A rabbit anti-mouse-Fc modified CM5rg sensor chip was used to bind the mAb, and scFv425-SNAP was passed through the system in HBS-EP buffer (Biacore; GE Healthcare) at concentrations of 5 μM to 20.57 nM and at a constant flow rate of 30 μL/minutes.
Results
SNAP- and CLIP-sequences were aligned, and the region with the highest diversity was chosen to derive the SNAP-specific peptide (Fig. 1). This region showed the greatest potential to yield mAbs that discriminate between SNAP and CLIP. The SNAP-specific peptide was coupled to KLH to increase the immune response in mice. When the antibody titer reached the range 1:51,200–201:102,400, the mouse was sacrificed and hybridoma technology was used to generate a SNAP-specific mAb (anti-SNAP-11), which could be purified and initially characterized by SDS-PAGE and western blot. Anti-SNAP-11 was purified with a total yield of 4.27 mg/L and the binding properties were investigated in detail.

Alignment of the SNAP and CLIP sequences showing the 18-residue peptide with the greatest diversity used for immunization.
ELISA experiments confirmed that anti-SNAP-11 recognizes various SNAP-tagged proteins (including those containing the original SNAP-tag, as well as the SNAPf variant), but does not bind to CLIP-tagged proteins (Fig. 2 and Supplementary Fig. S1). We found that 70 ng of anti-SNAP-11 was sufficient to achieve clear results, that is, four to five time fold higher than the background signal. We also found that 8 ng of the mAb was sufficient to detect the pure SNAP protein. The detection limit of the mAb was tested using serial dilutions, confirming that as little as 10 ng of each SNAP fusion protein was detectable (data not shown).
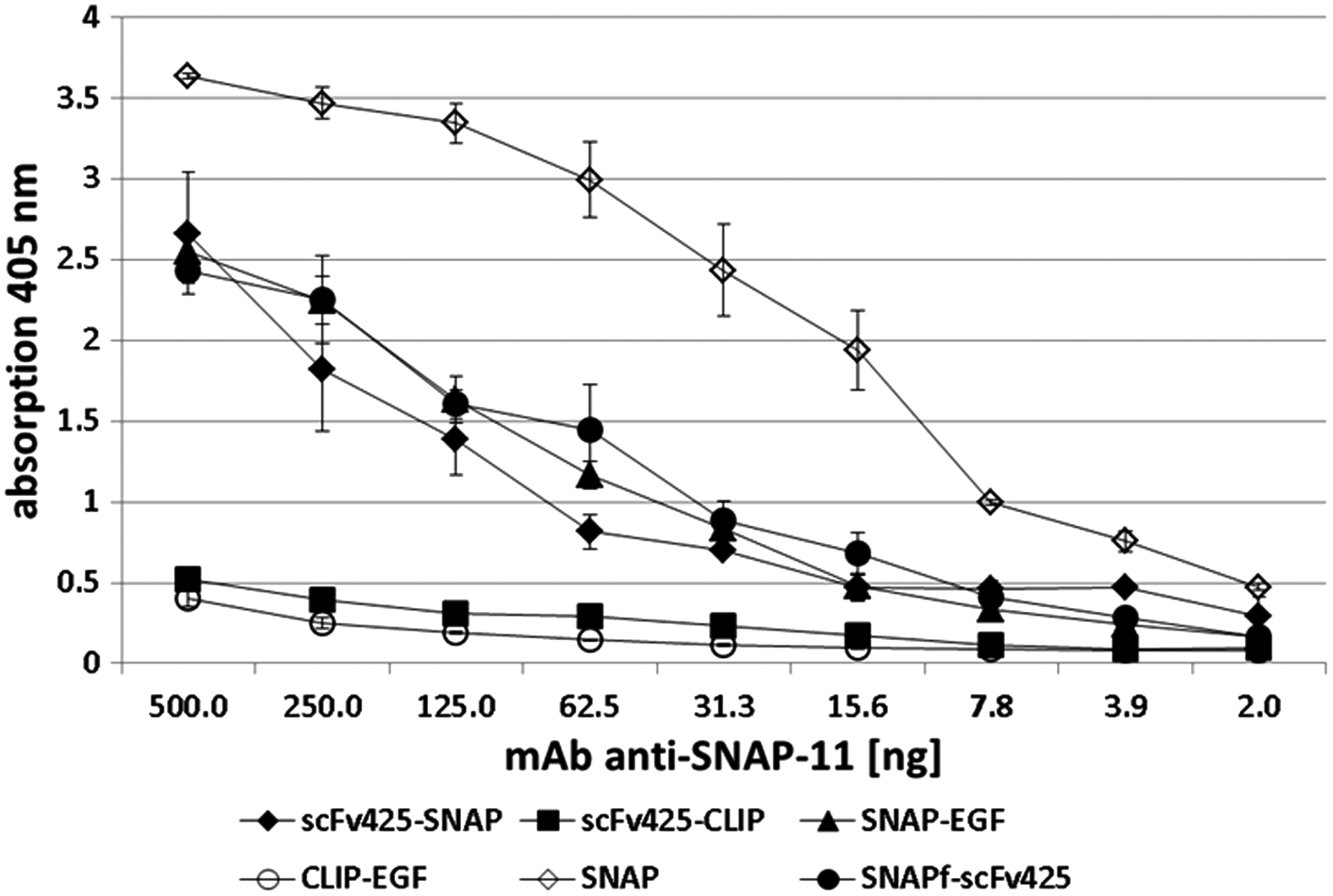
Detection of different SNAP/CLIP fusion proteins with anti-SNAP-11. A high protein binding microtiter plate was coated with 1 ng/μL of each protein, blocked with 1% BSA (w/v), and incubated with different amounts of anti-SNAP-11. Anti-SNAP-11 bound to SNAP and all SNAP fusions, but not the CLIP fusions, in a concentration-dependent manner. Error bars are representing standard deviations resulting from experiments performed in triplicates (n = 2).
Detection of denatured epitopes was confirmed by western blots with SNAP- and CLIP-tagged proteins (Fig. 3A). The detection limit of anti-SNAP-11 in western blots was ∼5 ng (Fig. 3B), which is similar to mAb M2D11.(18) The binding of anti-SNAP-11 to BG-coupled SNAP fusion proteins was also tested to ensure that the presence of BG-modified dyes did not affect the interaction between SNAP and the mAb. For this reason, scFv425-SNAP was coupled to BG-Biotin (NEB) and detected on a western blot by incubating with anti-SNAP-11 followed by GaM-AP and NBT/BCIP substrate (Fig. 3C). As a control for the successful coupling of BG-Biotin, the blots were developed with streptavidin-PO and DAB substrate (Fig. 3D). In both cases, a strong scFv425-SNAP signal was observed, confirming that the labeling of SNAP-tagged proteins with BG fluorophores did not prevent the additional binding of anti-SNAP-11.
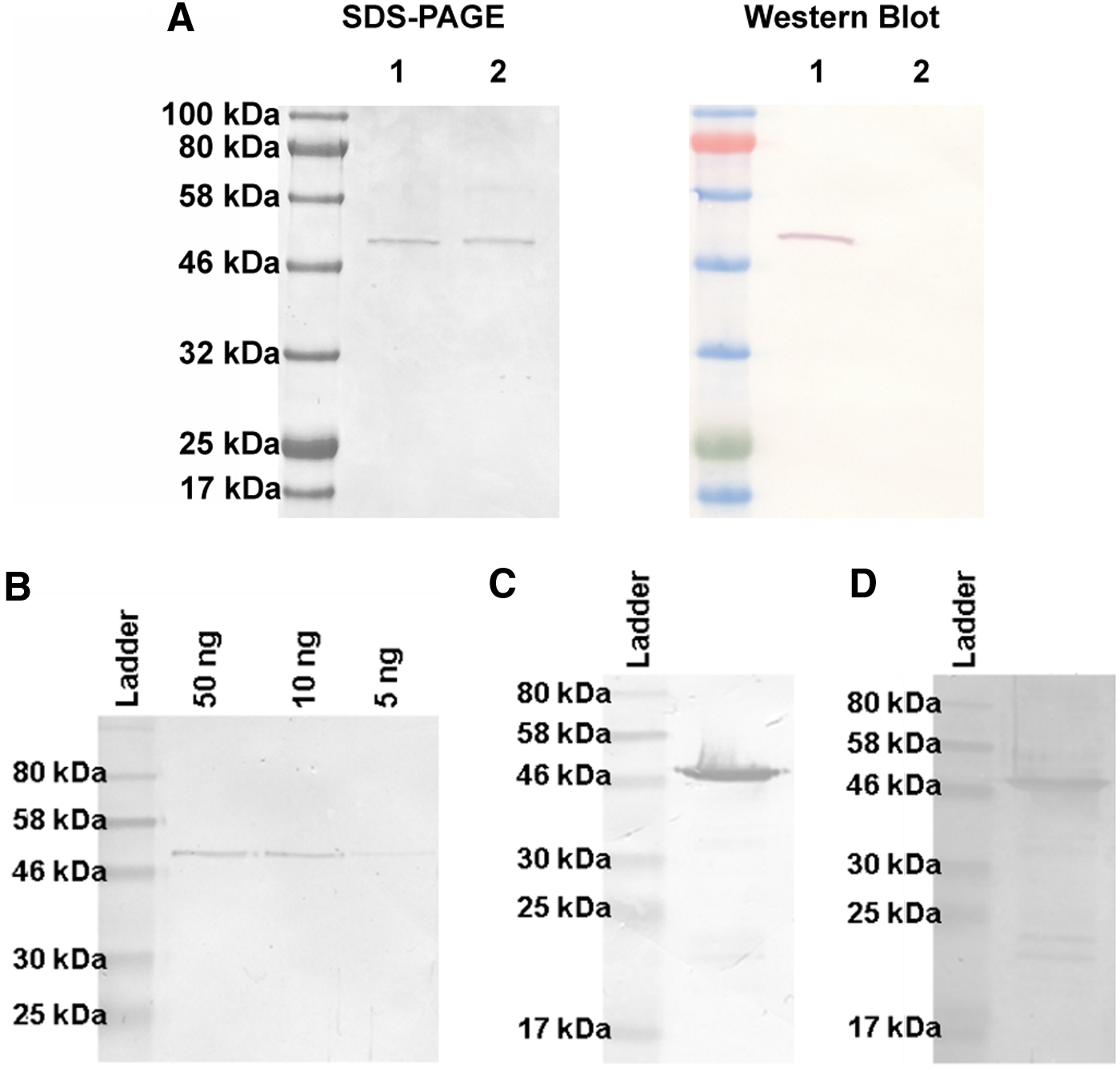
Detection of SNAP, BG-coupled SNAP, and CLIP fusion proteins by anti-SNAP-11 in western blot experiments.
Next, we compared the interaction between living cells and the mAbs anti-SNAP-11 and M2D11 by flow cytometry, using EGFR-positive cells, which are recognized by scFv425. Flow cytometry data confirmed that anti-SNAP-11 recognized scFv425-SNAP bound to the cell surface (Fig. 4A), whereas it was unable to recognize scFv425-CLIP bound to the cell surface, nor the cell surface itself in the absence of a SNAP-tagged protein. Under the same conditions, M2D11 recognized both scFv425-SNAP and scFv425-CLIP (Fig. 4B).
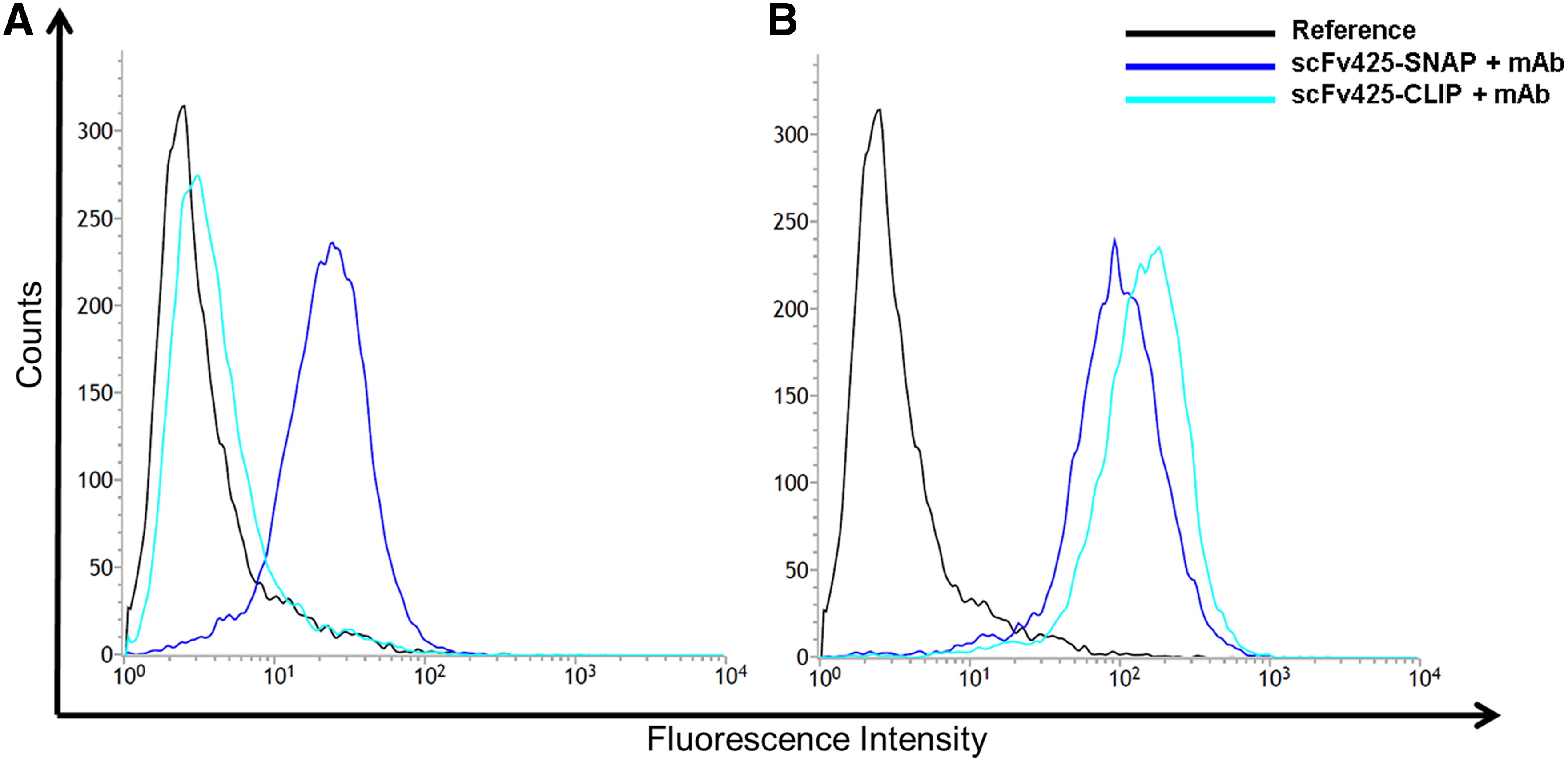
Flow cytometry to show specific binding of 50 ng anti-SNAP-11 or M2D11 to 3.5 × 105 MDA-MB-468 cells (ATCC: HTB-132) treated with scFv425-SNAP and scFv425-CLIP. (
The suitability of anti-SNAP-11 for immunohistochemistry was investigated using HER2-positive RD-derived tumors and an in-house anti-HER2scFv-SNAP protein. Anti-SNAP-11 also showed specific binding to RD-derived tumors decorated with the anti-HER2scFv-SNAP (Fig. 5E) and the signal was identical to that produced by M2D11 (Fig. 5D).

Immunohistochemistry of RD-derived tumor tissues using anti-HER2scFv-SNAP and PO-labeled anti-SNAP mAbs.
The affinity constant of anti-SNAP-11 immobilized on a RαM-Fc modified sensor chip was determined by capturing the scFv425-SNAP protein and measuring their binding activities by surface plasmon resonance. The affinity constant of anti-SNAP-11 was 2.49 × 10−7 M. The properties and potential applications of anti-SNAP-11 are compared to M2D11 in Table 1.
Discussion
The SNAP-tag is a promising approach for the study of protein functions, because it can be coupled to different BG-modified molecules, including fluorophores.(13,26) This allows SNAP-tagged proteins to be visualized by fluorescence microscopy, for the localization and tracking of proteins and cell structures in vivo.(27) NEB provides a polyclonal antibody for this purpose, but mAbs are advantageous, because they recognize one specific epitope and their homogeneity ensures consistent performance across different assay platforms.(16,28) We developed the first mAb against the SNAP-tag (M2D11) and this mAb detects both the original SNAP-tag and its derivative the CLIP-tag.(18) To distinguish between these structures and to allow their specific detection in dual labeling studies, we have developed the mAb anti-SNAP-11, which was raised against an 18-residue peptide that is specific for the SNAP-tag. This peptide sequence showed the highest degree of diversity between the tags in a sequence alignment (seven substitutions), making it the most likely sequence to yield an antibody specific for the SNAP-tag. The use of a small linear peptide from the surface of the folded structure also ensured that the antibody recognized both the native SNAP-tag in vivo and also the denatured SNAP-tag in procedures such as western blots.
We tested the specificity of anti-SNAP-11 using different ELISA and western blot experiments and found that the antibody recognized the original SNAP-tag and also the variant SNAPf in both native and denatured forms, but was unable to recognize the CLIP-tag. The ELISA detection limit for SNAP-tagged fusion proteins is 20 ng using mAb M2D11, but this was reduced to 10 ng using anti-SNAP-11. In western blots, the detection limit of both antibodies was similar (∼5 ng) compared to 10 ng reported for the VHH mAb derived from a llama library, although the experimental setups were different and not directly comparable. The coupling of BG-labeled molecules to the SNAP-tagged fusion proteins did not affect the binding of anti-SNAP-11, confirming that the simultaneous independent labeling of SNAP-tagged fusion proteins with BG dyes and anti-SNAP-11 is possible.
Flow cytometry and immunohistochemistry are commonly used diagnostic methods, so it was important to verify that anti-SNAP-11 is suitable for these procedures.(29,30) As previously reported for M2D11, anti-SNAP-11 was able to label cell structures clearly and specifically in both techniques, but the ability to selectively label SNAP-tagged proteins without labeling CLIP-tagged proteins represents a substantial improvement. In the flow cytometry experiments, M2D11 yielded a stronger signal than anti-SNAP-11 when each mAb was used to detect scFv425-SNAP bound to the surface of EGFR-positive cells. The affinity constant of anti-SNAP-11 was 2.47 × 10−7 M as determined by SPR spectroscopy, which is a midrange value compared to other murine mAbs.(31) The affinity of M2D11 was threefold higher (8.2 × 10−8 M), although this was determined using a different antigen (SNAP rather than scFv425-SNAP) so direct comparison is not possible. However, the lower affinity of SNAP-11 might be one explanation for the difference in signal strength detected in flow cytometry.
In the future, anti-SNAP-11 could be coupled to Sepharose beads and used for the immunoprecipitation or purification of SNAP-tagged fusion proteins, analogous to the use of mAbs recognizing, for example, Influenza virus hemagglutinin to precipitate proteins containing a hemagglutinin-tag,(32) thus providing an alternative to the widely used His6-tag. Compared to the c-myc-tag and the corresponding mAb 9E10, the affinity constant of 5.6 × 10−7 is nearly in the same range as our new mAb.(33) The commonly used tags for purification and their affinity constants, for example, His6-tag to Ni2+ (1.4 × 10−8 M)(34) or Strep-tag to streptavidin (3.7 × 10−5 M)(35) imply the later application of anti-SNAP-11 for affinity chromatography.
Anti-SNAP-11 bound to RD-derived tumor tissues in immunohistochemical experiments when the primary antigen (HER2) was detected with anti-HER2scFv-SNAP. The weak brown staining in the negative controls (Fig. 5B, C) reflects the absence of a blocking step to inactivate endogenous peroxidases with H2O2, because this treatment would have destroyed the samples by detaching the sections from the slides.
For both flow cytometry and immunohistochemistry, double labeling would be possible if anti-SNAP-11 is combined with a CLIP-specific mAb, which could potentially be raised by immunizing mice with the equivalent CLIP-specific 18-residue peptide. Theoretically, triple staining would be possible by combining BG-modified and BC-modified dyes with anti-SNAP-11 to monitor the behavior of three different surface proteins simultaneously. However, the use of fluorophores for live cell imaging is challenging because photobleaching can affect results based on fluorescence intensity.(11) The anti-SNAP-11 mAb helps to address this issue by allowing the quantitative staining of cells and tissues in a manner that is unaffected by exposure.
Conclusions
We have generated a murine mAb anti-SNAP-11, which is specific for SNAP-tagged but not CLIP-tagged proteins. Anti-SNAP-11 recognizes native and denatured epitopes of the SNAP variants by ELISA, western blot, flow cytometry, and immunohistochemistry, but the CLIP-tag is not recognized. In addition, the SNAPf variant was detected by anti-SNAP-11 in ELISA. This novel antibody is suitable for a variety of immunological methods and can therefore broaden the applications of the SNAP-tag technology.
Footnotes
Acknowledgments
The authors thank Kai Fuhrmann and Severin Schmies for hybridoma cell culture work, Holger Spiegel and Markus Sack (Fraunhofer IME) for SPR spectroscopy, and Hannes Brehm for the HER2-positive mouse tumor used for IHC. The work was funded by the INTERREG IV A project Microbiome (Grant No. EMR.INT4-1.2.-2009-11/058): “Avec le soutien du Fonds Européen de Développement Régional–La Commission Européenne investit dans vorte avenir. Me de steun van et Europees Fonds vorr Regionale Ontwikkeling–De Europese Commissie investeert in uw toekomst. Gefördert durch den Europäischen Fonds für Regionale Entwicklung–Die Europäische Kommission investiert in Ihre Zukunft.”
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.