
Abstract
More recently, alternative splicing of specific genes are investigated for their therapeutic potential. In particular, we reported the existence of BCR-ABL alternative splicing isoforms, in about 80% of Philadelphia-positive patients, which lead to the expression of aberrant proteins. These fusion proteins are characterized by an orphan initial and correct Bcr portion attached to a 112 amino acid sequence, arising from the impairment in the reading frame (reading of ABL exon 4 and 5). We demonstrated that these Abl-out-of-frame (OOF) isoforms could have an immunological role with therapeutic implications. The aim of this study was to characterize a new monoclonal antibody (MAb) specific for Abl-OOF protein portion, for diagnostic use, to detect this biomarker in Philadelphia chromosome-positive chronic myelogenous leukemia (CML) patients and to generate novel approaches in the immunotherapy. 5F11G11 MAb recognizes the OOF protein portion of the native full-length Bcr/Abl-OOF protein expressed in cells transiently transfected, as demonstrated by immunoprecipitation and immunofluorescence. In addition, we demonstrate the MAb's ability to recognize the alternative hybrid Bcr/Abl fusion protein expressed in leukemic cells from CML patients, to support the possible use of 5F11G11 MAb as a diagnostic tool to select patients with Philadelphia chromosome-positive CML that could be eligible for an immunotherapeutic approach with this new antigen.
Introduction
R
The Abl-OOF portion was largely studied for its attractive immunological function,(3–5) and it confirmed the presence of OOF-peptide-specific T cells in the peripheral blood of CML patients and their ability to lyse primary autologous CML cells, after in vitro sensitization.(1) The importance of OOF fusion proteins as new leukemia antigens for vaccination strategies was confirmed in vivo as well as in HLA-A2.1 transgenic mice model.(2)
These results led us to propose Abl-OOF portion as possible candidates for CML-specific tumor antigen for use in attempts at complete eradication of leukemic clones. In addition, we also showed that the Bcr/Abl-OOF protein was able to increase proliferation, migration, and to inhibit apoptosis, demonstrating that alternative proteins could have additional biological properties in tumorigenesis.(6) Therefore, Bcr/Abl-OOF is actively involved in the signaling of the product of the Ph chromosome by its oncogenic properties and could provide an additional target for developing new therapies to eradicate CML stem cells. This speculation could have tremendous consequences from a therapeutic standpoint.
To have new tools to study the specific tumor antigen Bcr/Abl-OOF, we produced and characterized a monoclonal antibody (MAb) specific for a portion of OOF amino acidic sequence.(3) In this study, we characterized a new MAb specific for Abl-OOF protein portion, to provide additional instruments for a diagnostic use, to detect this biomarker in Ph chromosome-positive CML or ALL patients and to generate novel approaches in the immunotherapy.
Materials and Methods
Synthesis of Abl-OOF portion
Recombinant Abl-OOF protein portion was obtained as previously described.(1) To obtain the purified 112-amino acid OOF Abl portion, the MBP-OOF fusion protein was cleaved by 50 U/g endoprotease Kex at room temperature for 2 hours. The resulting protein solution was loaded onto an amylose resin column and purified by affinity chromatography.
Immunization protocol
CD2F1mice (Charles River), 7–12 weeks old, were immunized subcutaneously four times at 2-week intervals with 50 μg/mouse of recombinant Abl-OOF portion. The antigen was emulsified in the same volume (100 μL/mouse) of Incomplete Freund's Adjuvant (Sigma-Aldrich, St. Louis, MO) and injected subcutaneously, followed by three booster injections at 2-week intervals. Blood from the mice was collected for sera 5 days after the last immunization. Test bleeds were assayed for positive reactions to indirect enzyme-linked immunosorbent assay (ELISA). Mice were housed in appropriate animal care facilities and handled according to international guidelines for experiments with animals.
Generation of OOF hybridomas
Spleen cells from immunized mice were fused to P3 × 63Ag8.653 mouse myeloma cells (Biological Bank, Istituto Nazionale per la Ricerca sul Cancro IST, Genova, Italy) in the presence of a 50% solution (wt/mL) of polyethylene glycol (Molecular Weight 3350; Sigma-Aldrich) to produce MAbs according to standard procedures.(7) Cells were plated in 96-well plates (Corning-Costar Corp) and cultured at 37°C in a humidified atmosphere in the presence of 5% CO2 and 95% air in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS),
Selected hybridoma lines were later grown, and concentrated MAb supernatants were purified using Protein A IgG Purification Kit (Thermo Scientific Pierce, Rockford, IL), according to the manufacturer's instructions.
ELISA assay
ELISA 96-well plates (Immobilizer–Nunc Thermo Scientific) were coated with 1 μg/well of peptides or the 112 Abl-OOF amino acidic portion diluted in phosphate-buffered saline (PBS) at 4°C overnight. Plates were washed three times with PBS and blocked with 5% bovine serum albumin (BSA) (Sigma-Aldrich) in PBS at 37°C for 1 hour. Supernatants (50 μL) from hybridoma cultures, immune serum, or purified antibody diluted in PBS +0.1% BSA were added and incubated at 37°C for 2 hours. Plates were washed three times with PBS +0.2% Tween-20 (Sigma-Aldrich) before incubation with secondary goat anti-mouse IgG antibody conjugated to alkaline phosphatase (Millipore, Bedford, MA) at 37°C for 1 hour.
After another three washes with PBS +0.2% Tween-20, the plates were incubated with p-nitrophenyl phosphate (Sigma-Aldrich) diluted to 1 mg/mL in diethanolamine substrate buffer (Thermo Scientific, Waltham, MA) at 37°C for 30 minutes. The optical density at 405 nm was detected on a Victor3 plate reader (Perkin Elmer, Waltham, MA). Isotypes of the MAb were determined using the Mouse Mono-Ab-ID kit (Invitrogen, Carlsbad, CA) through the capture assay according to the manufacturer's instructions.
Cell culture, transfection for tumor antigen expression
293T cell line was maintained in RPMI 1640 (Euroclone) supplemented with 10% heat-inactivated FCS (Sigma-Aldrich) and 2 mM glutamine (Euroclone) at 37°C in a 5% CO2 humidified atmosphere. Transient transfections were performed using Fugene HD transfection reagent (Roche Diagnostic S.p.A, Basel, Switzerland) according to the manufacturer's instructions. The pcDNA3.1-BCR/ABL-OOF construction was previously described.(3) To enrich the percentage of transfected cells, 2 days after, the transfection stable clones were selected with G418 (100 μg/mL) and then expanded.
Healthy human donors and patients
Human bone marrow specimens were collected from healthy donors or chronic myeloid leukemia patient at diagnosis following written informed consent.
Immunofluorescence assay
To analyze the antibody's ability to recognize the Bcr/Abl-OOF protein in bone marrow cells from CML patients or healthy donors, cells were permeabilized after fixation for 5 minutes and blocked for 45 minutes with 10% BSA in PBS. Cells were incubated for 2 hours with 5 μg/mL anti-OOF Mab, and the binding was detected by 30 minutes incubation with secondary goat anti-mouse Alexa Fluor 488 IgG antibody (Molecular probes) diluted 1:1000 in PBS. Cells were then treated for 5 minutes with red propidium (Sigma-Aldrich) for nuclear staining and analyzed by confocal scanning microscope (LSM 510; Carl Zeiss MicroImaging Inc., Thornwood, NY) using 63 × objectives.
Cytofluorimetric analysis
Bone marrow cells from CML patients or healthy donors were permeabilized after fixation for 5 minutes and blocked with FCS at room temperature for 30 minutes. 1 × 106 cells were then incubated on ice for 1 hour with MAb 5F11G11 diluted in washing buffer (PBS containing 5% FCS and 0.1% sodium azide, 100 μL/sample) (2 μg/mL). After three washes in washing buffer, cells were stained with phycoerythrin-conjugated anti-mouse Ig (Dako, Denmark A/S, Glostrup, Denmark) diluted 1:10 in washing buffer. Incubation was carried out in the dark at 4°C for 30 minutes followed by three washes in FACS buffer. Isotype-matched nonspecific antibodies were used as control (Dako). Cells were analyzed by FACSCalibur (Becton Dickinson), and BD Cell Quest Pro software (Becton Dickinson) was used to analyze data.
For all samples, 50,000 events were acquired in the R1 region gate, which was defined based on forward and side light scatter properties to exclude debris.
Western blot and immunoprecipitation
Recombinant Abl-OOF protein portion was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to 0.45 μm nitrocellulose membranes (Invitrogen) at 4°C overnight. The blots were then blocked with PBS +0.1% Tween +3% skim milk at room temperature for 90 minutes. After washing three times with PBS +0.1% Tween and twice with distillate water, the blots were incubated at room temperature for 1 hour with blocking solution containing 1.5 or 3 μg/mL MAb 5F11G11. As negative control, blot was incubated with only the secondary antibody. As an additional control, an uncorrelated recombinant protein with similar molecular weight, tumor growth factor-alpha (eBioscience), was used and blotted with 3 μg/mL 5F11G11. The membranes were washed three times with PBS +0.1% Tween and incubated with goat anti-mouse IgG-conjugated to horseradish peroxidase (Thermo Scientific-Pierce) at room temperature for 45 minutes. Following three washes with PBS +0.1% Tween and two with distillate water, the membranes were incubated with the Protein Detection System (Genespin s.r.l. Milan, Italy).
For immunoprecipitation experiments, cells were lysed with lysis buffer containing 50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM Na3VO4, 25 mM NaF, protease inhibition cocktail (all Sigma-Aldrich), and 1 mM ethylenediaminetetraacetic acid. Lysates were clarified by centrifugation at 15,900 g at 4°C for 30 minutes. The BCA Assay Reagent Kit (Thermo Scientific-Pierce) was used to determine protein concentrations.
Clarified lysates were incubated with 5 μg/mL MAb 5F11G11 at 4°C overnight and then bound to protein G agarose (Sigma-Aldrich, St. Louise, MO) at 4°C for 1 hour. The agarose was washed five times with lysis buffer, and proteins were released from the agarose by boiling in NuPAGE LDS sample buffer (Invitrogen) for 5 minutes. The proteins were then subjected to SDS-PAGE and transferred to 0.45 μm nitrocellulose membranes (Invitrogen) at 4°C overnight. After washing three times with PBS +0.1% Tween and twice with distillate water, the blots were incubated at room temperature for 1 hour with purified rabbit polyclonal antibody specific for Bcr (7 μg/mL) (Santa Cruz Biotechnology, Dallas, TX) diluted in blocking solution. The membranes were washed three times with PBS +0.1% Tween and incubated with goat anti-rabbit Ig conjugated to horseradish peroxidase (Thermo Scientific-Pierce) at room temperature for 45 minutes. Following three washes with PBS +0.1% Tween and two with distillate water, the membranes were incubated with the Protein Detection System (Genespin s.r.l.).
In-cell western assay quantitative immunofluorescence assay
Cells were cultured overnight in 96-well plates in complete medium culture, and then they were fixed with 3.7% formaldehyde for 20 minutes, washed with PBS, and then permeabilized with Triton X-100 (0.1% in PBS) for 15 minutes. Cells were blocked with Odyssey blocking buffer (LI-COR Bioscience) for 90 minutes at room temperature. After incubation in a humid chamber overnight at 4°C with MAb 5F11G11 diluted in Odyssey blocking buffer, cells were washed with PBS +0.1% Tween-20 and incubated for 1 hour at room temperature with LI-COR IRDye 800 labeled secondary antibodies (1:800) and with CellTag 700 (1:500) for cell number normalization, diluted in Odyssey blocking buffer. CellTag700 is a nonspecific cell stain that accumulates in both the nucleus and cytoplasm. Finally, cells were washed five times with PBS +0.1% Tween-20 before the scan of the plate by Odyssey Infrared Imaging System (LI-COR Bioscience). Data were obtained from quadruplicate experiments.
RNA extraction and quantitative real-time polymerase chain reaction
To amplify alternative splice variants, 1 μg of total cellular RNAs, extracted from bone marrow cells of CML patients at diagnosis, using the guanidium/thiocyanate-phenol/chloroform method,(8) was reverse transcripted in c-DNA using random examers. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed as previously described(1) and the amount of the BCR/ABL-OOF alternative transcripts, normalized for ABL1 as control gene, was expressed as relative quantification using the equation 2−ΔΔCt, following calibration with a pool of normal samples (healthy donors) that do not express our genes of interest.
Statistical analysis
Statistical significance between groups was determined by one-way ANOVA; p < 0.05 was considered significant. All calculations were performed using GraphPad Prism 3.0 (GraphPad Software LaJolla, CA).
Results
In this study, we characterized a new MAb specific for the Abl-OOF sequence (Fig. 1). In the past, we obtained a MAb specific for a portion of this sequence (peptide 22–53), immunizing C57 BL/6 mice transgenic for the human leukocyte antigen class I A2.1 molecule.(3) Our results indicated that only the 22–53 sequence was immunogenic in this mouse strain and was able to produce MAb specific for Abl-OOF portion. To obtain new specific MAbs for Abl-OOF amino acidic sequence, to provide additional instruments for a diagnostic use, we immunized CD2F1 mice, a cross between female BALB/c and male DBA/2, with the complete sequence of Abl-OOF portion (Fig. 1), as described in the Materials and Methods section.

Synthetic Abl-OOF amino acidic sequence and peptides. The 112 Abl-OOF amino acidic sequence and sequence of 4 peptides, characterized by easy synthesis, purification, and stability. OOF, out of frame.
All mice developed antibodies specific for OOF protein portion (Fig. 2A), in particular, ELISA assay revealed the immunogenicity of the entire Abl-OOF amino acidic sequence in this mouse strain (Fig. 2B), as proved by the presence of antibodies specific for the peptides 1–20, 22–53, 32mer, and 29mer, each corresponding to a portion of the Abl-OOF sequence (Fig. 1).
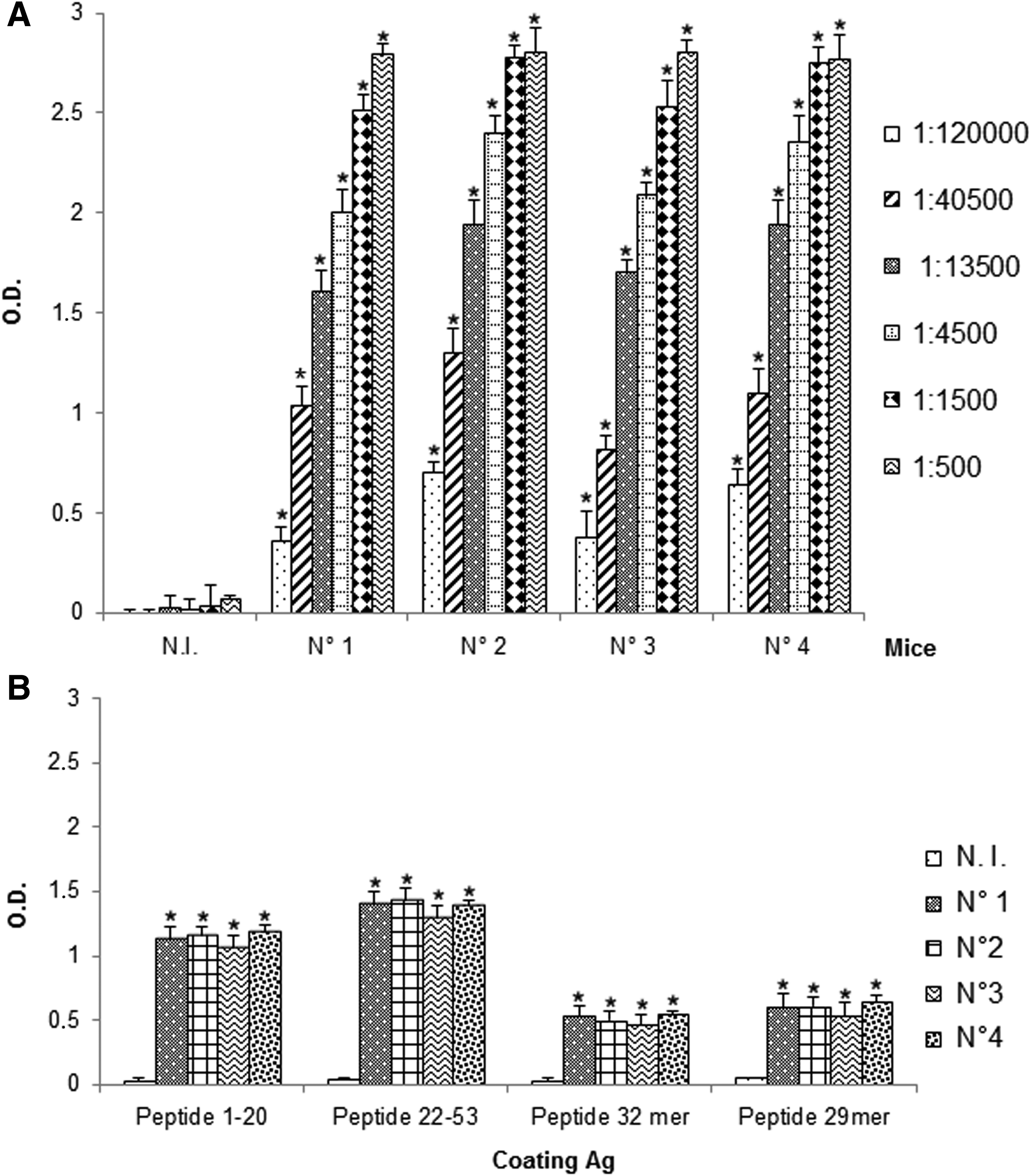
Antibody detection in sera from mice immunized with Abl-OOF protein portion. The presence of specific antibodies for Abl-OOF sequence
Spleens from immunized mice were fused to P3 × 63Ag8.653 myeloma cells to develop anti-OOF MAbs. ELISA was used to screen hybridoma culture supernatants for binding activity to the 112 Abl-OOF portion. After cloning by limiting dilution, we obtained stable hybridomas, and we selected the 5F11G11 clone, able to bind peptide 32mer in a concentration-dependent manner (Fig. 3). The isotope analysis indicated that this MAb was of the IgG1 subtype, λ-chain (data not shown).
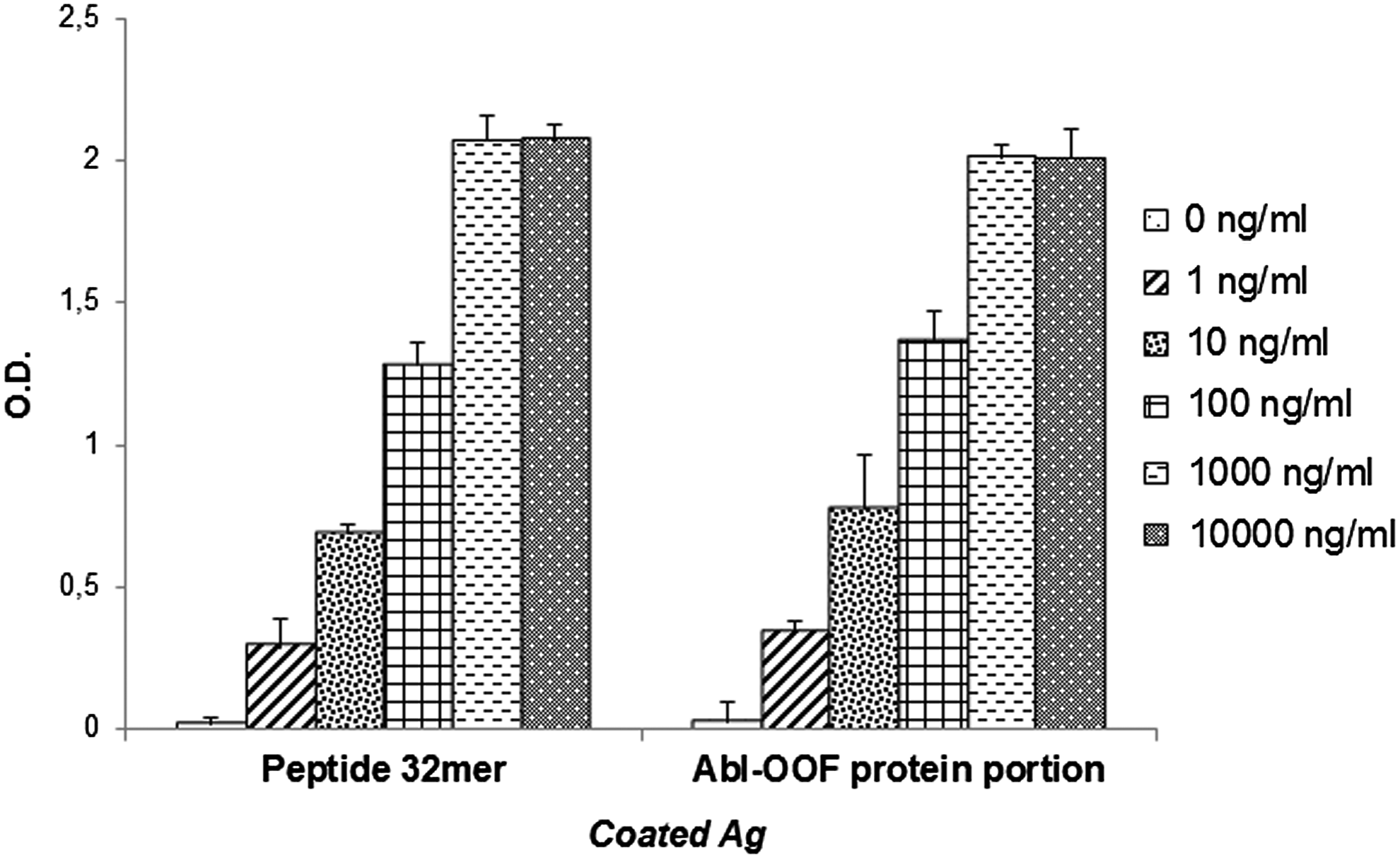
Specific binding analysis of MAb 5F11G11 for peptide 32mer and for recombinant OOF Abl portion by ELISA. Peptide or Abl-OOF portion were coated into microtiter plates, and serial dilutions of MAb were added. The binding of MAb to antigens was detected with horseradish peroxidase-conjugated goat anti-mouse IgG. “0 ng/nL”: negative control performed in absence of MAb 5F11G11. Results are expressed as mean ± standard error of three experiments. MAb, monoclonal antibody.
Specific binding of MAb 5F11G11 was confirmed by western blot analysis (Fig. 4). In fact, it was able to bind to the recombinant Abl-OOF portion, corresponding to a 12-kDa protein band.
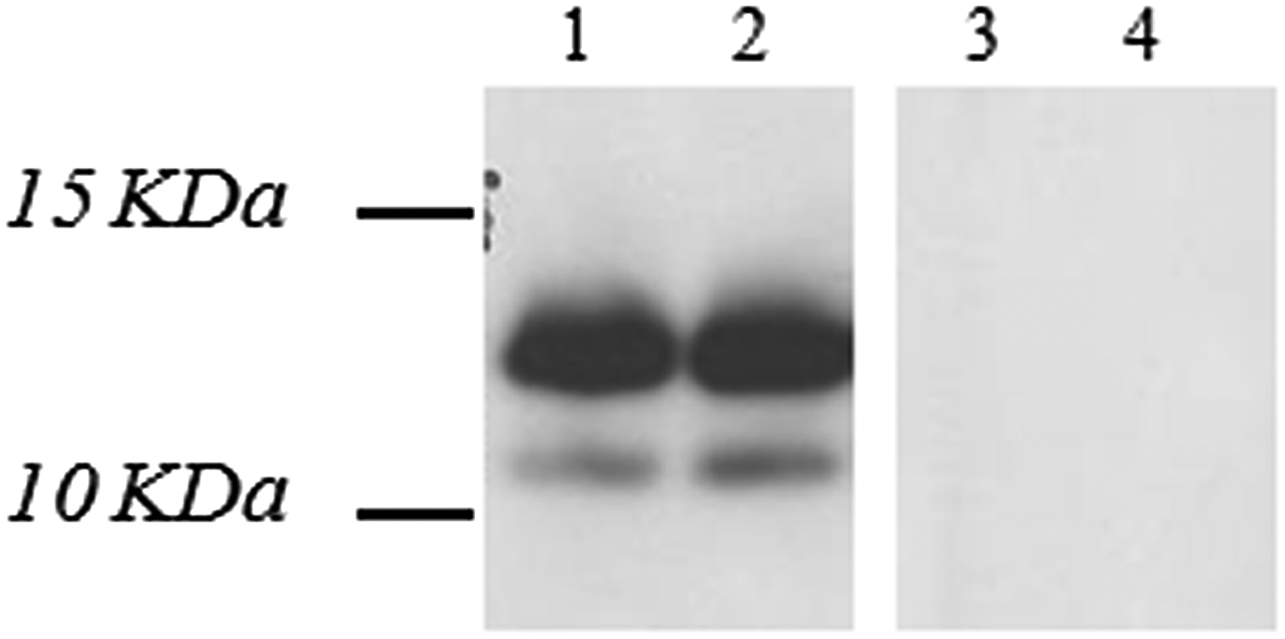
Specific binding analysis of MAb 5F11G11 for Abl-OOF by western blot. One hundred twelve Abl-OOF portion (100 ng/well) was resolved on SDS-PAGE under reducing conditions. The resolved gel was transferred to a blotting membrane and incubated with MAb 5F11G11 at the concentration of 1.5 and 3 μg/mL (lane 1 and lane 2, respectively). Bound antibody was detected with an horseradish peroxidase-conjugated goat anti-mouse IgG. As negative control, blot was incubated with the secondary antibody, without MAb 5F11G11 (lane 3). In addition, an uncorrelated recombinant protein with similar molecular weight, TNF-alpha (17 kDa), was used (100 ng/well) and blotted with 3 μg/mL 5F11G11 (lane 4). Sizes (kDa) of molecular mass markers are indicated. Abl-OOF portion: 12 kDa. SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; TNF, tumor growth factor.
To evaluate the ability of the MAb to recognize the native state of alternative Bcr/Abl-OOF protein, we used 293T cells transfected with the expression vector pcDNA 3.1 BCR/ABL-OOF, generated by our laboratory as previously described.(2) Forty-eight hours after transfection, 293T cells were subjected to in-cell western assay and immunoprecipitation.
“In-cell western” is an immunocytochemical assay, in which the cells were grown in multiwell plates, fixed, and stained directly with the specific antibody, to detect the intracellular target. MAb 5F11G11 was able to recognize Bcr/Abl OOF expressed in 293T cells transfected, and the absence of signal on cells transfected with the empty vector confirmed the specificity of the binding (Fig. 5).
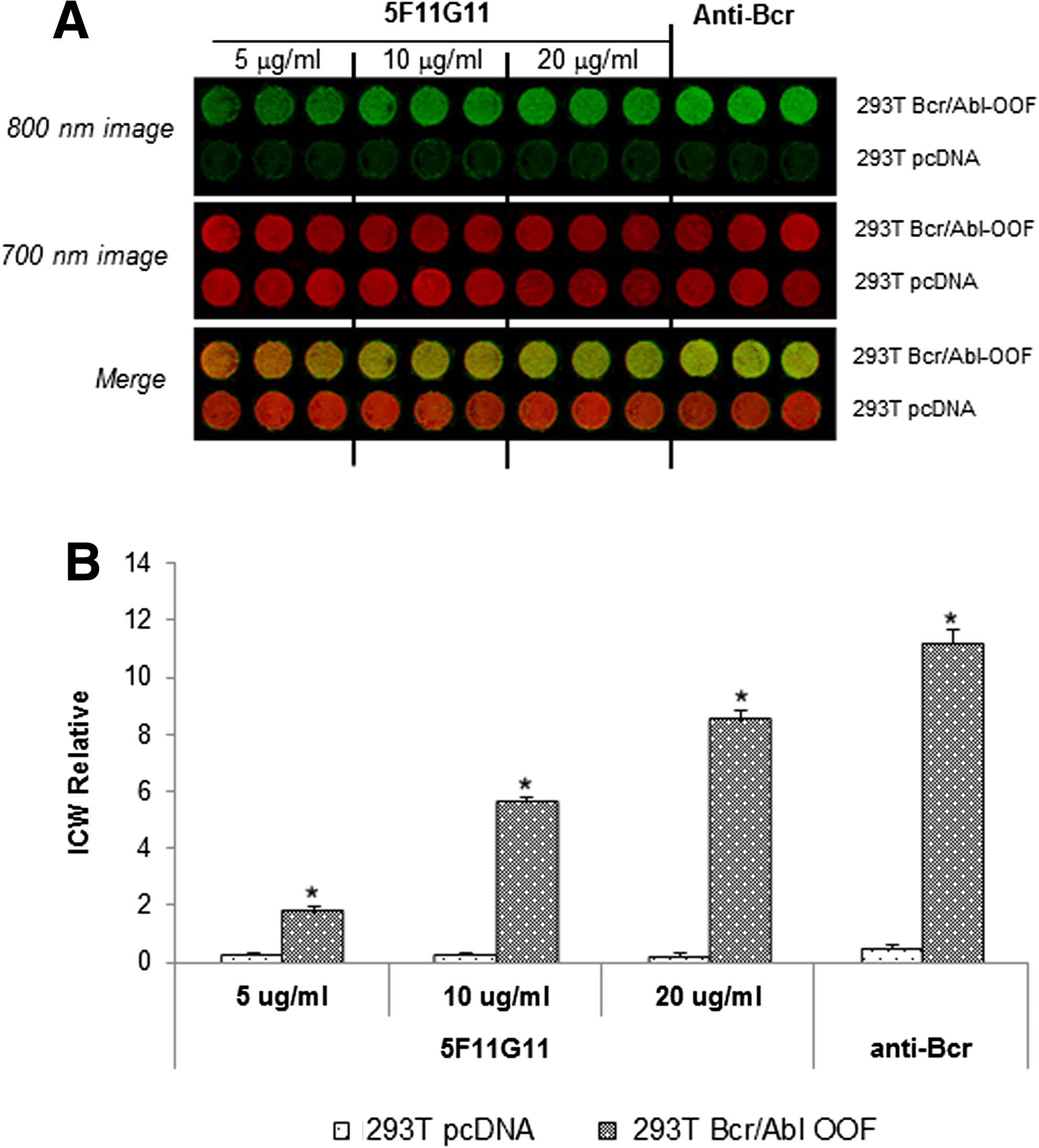
In-cell western assay on BCR/ABL-OOF transfected 293T cells. Cells transiently transfected with pcDNA3-1-BCR/ABL-OOF (293T Bcr/Abl-OOF) or with pcDNA3.1 empty vector (293T pcDNA) were fixed, permeabilized, and probed with MAb 5F11G11 (5, 10, and 20 μg/mL) or anti-Bcr polyclonal antibody (7 μg/mL).
At the same time, 293T cells transfected with the expression vector pcDNA 3.1 BCR/ABL-OOF were lysed and protein extracts were immunoprecipitated with MAb 5F11G11. An anti-Bcr-purified polyclonal antibody was used for immunoblotting analysis. As shown in Figure 6, we detected a 121-kDa band corresponding to the predicted size of the transfected full-length alternative Bcr/Abl-OOF fusion protein. This band was also present when we used a commercial anti-Bcr antibody as positive control. When lysates of 293T cells transfected with the empty vector were immunoprecipitated with MAb 5F11G11, we did not detect band to confirm the specificity of our MAb.

Immunoprecipitation of 293T Bcr/Abl-OOF cell lysates with MAb 5F11G11. Lysates from cells transiently transfected with pcDNA3-1-BCR/ABL-OOF or with pcDNA3.1 empty vector were immunoprecipitated with MAb, resolved, and transferred to nitrocellulose membranes. An anti-Bcr polyclonal antibody was used in western blot of the immunocomplexes, leading to the detection of a 121-kDa band corresponding to the predicted size of the full-length alternative Bcr/Abl-OOF fusion protein in 293T Bcr/Abl-OOF immunoprecipitates (lane 2). No corresponding band was visualized in 293T cells transfected with the empty vector (lane 3). As positive control 293T Bcr/Abl-OOF cell lysates were examined by immunoblotting with anti-Bcr polyclonal antibody (7 μg/mL) (lane 1).
Finally, we verified the possibility that MAb 5F11G11 was able to recognize the alternative antigen expressed in patients with BCR/ABL positive CML. Therefore, we analyzed flow cytometry bone marrow cells from CML patients at diagnosis, positive for BCR/ABL-OOF transcripts, along with bone marrow cells from healthy donors, as normal control. We chose three CML patients with different expression level of BCR/ABL-OOF transcript to a qRT-PCR analysis (Table 1). As shown in Figure 7, 5F11G11 antibody was able to detect a positive signal in all three patients.
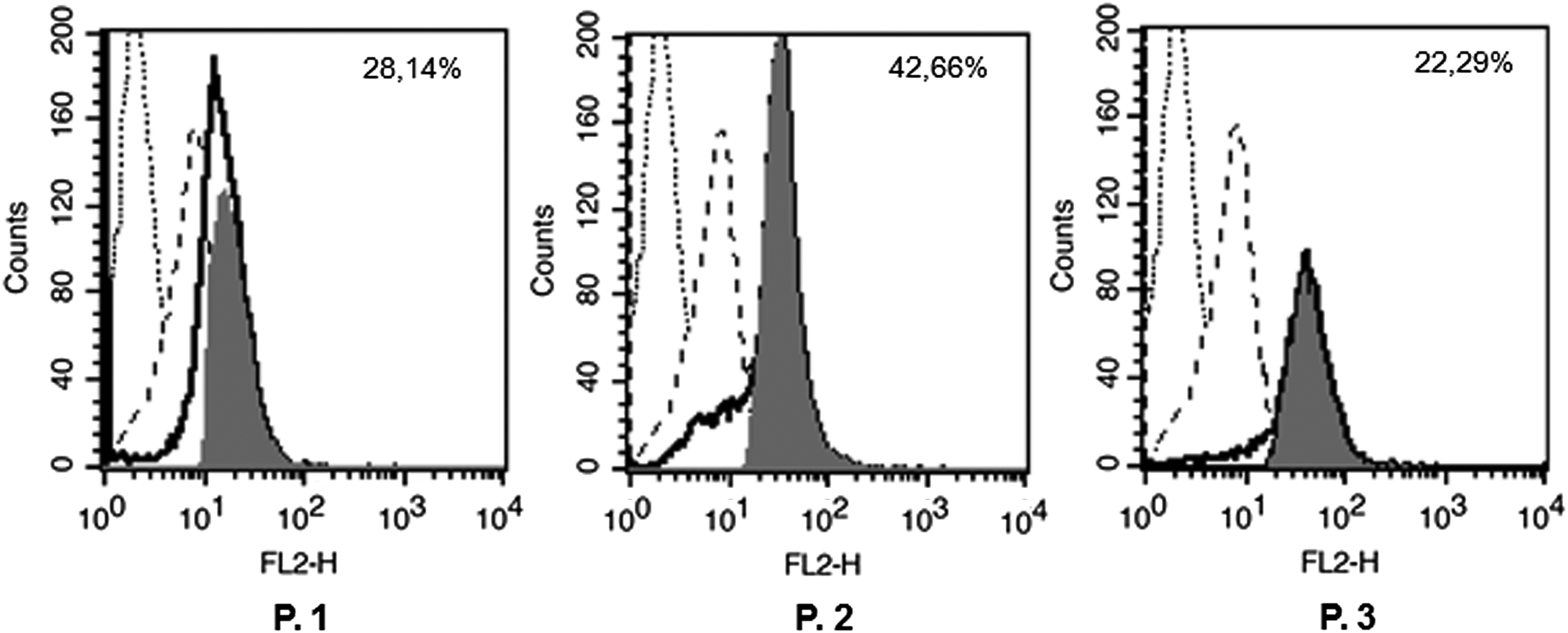
Binding analysis of MAb 5F11G11 to Bcr/Abl OOF-positive bone marrow cells by flow cytometry. Bone marrow cells from three CML patients (P. 1, P. 2, P. 3) were fixed, permeabilized and stained with MAb 5F11G11. After incubation with PE-conjugated secondary antibody, cells were analyzed by flow cytometry to assess the antibody binding to bone marrow leukemic cells. The empty histograms (heavy solid line) represent staining of 5F11G11on CML BM cells and the dashed line histograms represent staining on BM cells from a healthy donor. Histogram overlay subtraction analysis (filled histograms) was performed using BD Cell Quest Pro software, to calculate the percent of OOF-positive CML BM cells. For each patient is indicated the % value obtained. Fluorescence of isotype control antibody stained cells is indicated as dotted line histogram. CML, chronic myelogenous leukemia.
Bone marrow cells from CML patients at diagnosis were analyzed to evaluate the BCR/ABL-OOF alternative transcripts expression. The amount of BCR/ABL-OOF alternative transcripts was normalized for ABL1 as control gene, and expressed as relative quantification using the equation 2−ΔΔCt.
CML, chronic myelogenous leukemia; OOF, out of frame.
In parallel, we performed an immunofluorescence assay, and we confirmed the MAb's ability to recognize Bcr/Abl-OOF protein expressed in CML cells (Fig. 8B). In Abl-OOF negative bone marrow cells was observed a low background, due to the ability of some cells to uptake antibody in a nonspecific way (Fig. 8A). However, the antibody signal quantification suggested a significant difference between the two specimens (Fig. 8C).

Immunofluorescence assay on CML bone marrow patient. Fixed bone marrow cells from CML patient
Discussion
In these last years, research studies have improved our knowledge on CML molecular mechanism with the development of specific target therapies. Because BCR/ABL is necessary and sufficient for the leukemic phenotype, the generation of tyrosine kinase inhibitors (TKIs) represents the gold standard choice for all newly diagnosed CML patients. They have very low side effects and good tolerability.(9,10)
Unfortunately, cure of CML is not achieved by blocking Bcr/Abl kinase activity, and beside TKIs, a small percentage of patients develop resistance with consequent progression in accelerated phase and blast crisis.(11–13)
The only therapy able to efficiently eradicate and cure CML remains bone marrow transplantation.
Furthermore, it seems that myeloid leukemia cells are involved in immune system control and for this reason induction of immune-mediated response could be a good strategy.
In this scenario, it is important to develop new therapeutic options to help the immune system to react and to destroy cancer cells.(14,15)
In our laboratory tumor-specific Bcr/Abl-OOF antigen was largely studied for its attractive immunological function.(2–5) Our previous observations suggest the presence of immune surveillance against the Bcr/Abl-OOF fusion proteins in Ph-positive patients.(1) Indeed, Abl-OOF-specific cytotoxic T lymphocytes were revealed in the peripheral blood of CML patients, but not in healthy donors. These results indicate that the Abl-OOF specific-CTLs are elicited in response to leukemic cells. In addition, our in vivo studies performed in the HLA-A2.1 transgenic mice model demonstrate the Abl-OOF protein portion's capability to induce a specific and multiple immune responses.(2,4) All together, these observations suggest the possibility of developing a peptide-based vaccination with Abl-OOF, to evoke CML-specific tumor antigen immune responses in Bcr/Abl-OOF-positive leukemic patients.
In addition, Panuzzo et al. showed that Bcr/Abl-OOF protein was able to increase proliferation, migration, and inhibit apoptosis, demonstrating that alternative proteins could have additional biological properties in tumorigenesis.(6) This recent demonstration of a direct Bcr/Abl-OOF involvement in the Bcr/Abl signaling(6) supports the idea that it could be an additional target for developing a new strategy to eradicate leukemic cells.
To provide new instruments for a diagnostic and immunotherapeutic use in Ph chromosome-positive CML or ALL patients, we produced and characterized a MAb specific for Abl-OOF portion.(3) In this study, we characterized a second MAb specific for this antigen, able to bind a different epitope of Abl-OOF portion.
The new murine MAb, named 5F11G11, is able to recognize the recombinant antigen in ELISA and western blot assays, as well as the native Abl-OOF portion expressed in 293T cells transiently transfected with pcDNA3-1-BCR/ABL-OOF, as demonstrated by in-cell western and immunoprecipitation assays.
Immunofluorescence and flow cytometric analysis performed on leukemic cells from Ph chromosome-positive patients at diagnosis revealed a specific stain for 5F11G11 antibody. These results demonstrate the ability of MAb 5F11G11 to recognize the antigen expressed in these cells.
This study on CML patients is very interesting and suggests a good sensitivity of MAb 5F11G11. Indeed, the Bcr-Abl alternative isoforms are poorly expressed in CML cells(1) but, despite this, we detected a good specific signal on tumor cells.
All these data demonstrate that our MAb would allow the identification of the Bcr/Abl-OOF tumor antigen with simple procedures, and it should be a valid tool to identify patients eligible for a targeted therapy, such as vaccination, to associate with conventional therapy of Ph chromosome-positive leukemic patients in cytogenetic remission(2,4) and to eradicate the percentage of Bcr/Abl-positive cells that can persist and the end of gold standard therapy.
Footnotes
Acknowledgment
The study was supported by Fernando Santarelli Foundation. Moreover, this work was partially supported by Consorzio Milano Ricerche.
Author Disclosure Statement
No competing financial interests exist.