
Abstract
Podoplanin is expressed in many cancers, including oral cancers and brain tumors. The interaction between podoplanin and its receptor C-type lectin-like receptor 2 (CLEC-2) has been reported to be involved in cancer metastasis and tumor malignancy. We previously established many monoclonal antibodies (mAbs) against human podoplanin using the cancer-specific mAb (CasMab) technology. LpMab-23 (IgG1, kappa), one of the mouse anti-podoplanin mAbs, was shown to be a CasMab. However, we have not shown the usefulness of LpMab-23 for antibody therapy against podoplanin-expressing cancers. In this study, we first determined the minimum epitope of LpMab-23 and revealed that Gly54–Leu64 peptide, especially Gly54, Thr55, Ser56, Glu57, Asp58, Arg59, Tyr60, and Leu64 of podoplanin, is a critical epitope of LpMab-23. We further produced human–mouse chimeric LpMab-23 (chLpMab-23) and investigated whether chLpMab-23 exerts antibody-dependent cellular cytotoxicity (ADCC) and antitumor activity. In flow cytometry, chLpMab-23 showed high sensitivity against a podoplanin-expressing glioblastoma cell line, LN319, and an oral cancer cell line, HSC-2. chLpMab-23 also showed ADCC activity against podoplanin-expressing CHO cells (CHO/podoplanin). In xenograft models with HSC-2 and CHO/podoplanin, chLpMab-23 exerts antitumor activity using human natural killer cells, indicating that chLpMab-23 could be useful for antibody therapy against podoplanin-expressing cancers.
Introduction
P
Transforming growth factor-β (TGF-β) regulates many physiological events including tumorigenesis.(26) We previously found that TGF-β induced podoplanin in human fibrosarcoma HT1080 cells and enhanced the platelet aggregating ability of HT1080.(27) TGF-β type I receptor inhibitor (SB431542) and short hairpin Smad4 inhibited the podoplanin induction by TGF-β, suggesting that TGF-β is a physiological regulator of podoplanin in tumor cells. Recently, another group confirmed this phenomenon using in vivo analysis.(28) Podoplanin-mediated epithelial–mesenchymal transition (EMT) reportedly resulted in increased invasiveness and extravasation of tumor cells, and treatment of mice with a TGF-β-neutralizing antibody inhibited podoplanin-mediated hematogenous metastasis in vivo, indicating that podoplanin promoted hematogenous metastasis in part by releasing TGF-β from platelets that was essential for EMT of tumor cells.
Many anti-podoplanin monoclonal antibodies (mAbs) have been reported, and almost all of them, such as D2-40, 18H5, and NZ-1, react with the peptides of podoplanin.(19,29–35) We have used our original technology to produce anti-glycopeptide mAbs (GpMabs) against podoplanin.(36–40) One of these GpMabs, LpMab-2, was shown to be a cancer-specific mAb (CasMab).(36) LpMab-2 recognizes both an aberrant O-glycosylation and a Thr55–Leu64 peptide from podoplanin. Because LpMab-2 reacts with podoplanin-expressing cancer cells but not with normal cells, as shown by flow cytometry and immunohistochemistry, it is expected to be useful for molecular targeting therapy against podoplanin-expressing cancers.(41) Another anti-podoplanin CasMab is LpMab-23.(42)
In this study, we produced human–mouse chimeric LpMab-23 (chLpMab-23) and investigated whether chLpMab-23 exerts antibody-dependent cellular cytotoxicity (ADCC) and antitumor activity.
Materials and Methods
Cell lines
Chinese hamster ovary cell lines (CHO)-K1, LN229, and P3U1 were obtained from the American Type Culture Collection (Manassas, VA). HSC-2 was obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). The LN319 cell line was provided by Prof. Kazuhiko Mishima (Saitama Medical University, Saitama, Japan).(43) CHO-S was purchased from Thermo Fisher Scientific, Inc. (Waltham, MA). Core fucose-knockout CHO-S cells (PDIS-5) was established using CRISPR-Cas9 in our previous study.(41) CHO-K1 and LN229 cells were transfected with podoplanin plasmids using Lipofectamine 2000 (Thermo Fisher Scientific, Inc., Waltham, MA) according to the manufacturer's instructions.(36)
The CHO-K1, CHO-K1/podoplanin, and P3U1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium containing L-glutamine (Nacalai Tesque, Inc., Kyoto, Japan) and 10% heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific, Inc.). HSC-2 cells were cultured in Eagle's minimal essential medium (Nacalai Tesque, Inc.) or Dulbecco's modified Eagle's medium (DMEM) containing L-glutamine and 10% heat-inactivated FBS. The LN319, LN229, and LN229/podoplanin cells were cultured in DMEM containing L-glutamine and 10% heat-inactivated FBS. Cells were kept at 37°C in a humidified atmosphere containing 5% CO2. All media contained 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 25 μg/mL of amphotericin B (Nacalai Tesque, Inc.).
Antibodies
LpMab-23, a mouse anti-podoplanin mAb (IgG1, kappa), was developed as previously described.(42) Human IgG was purchased from Beckman Coulter, Inc. (Fullerton, CA) or Sigma-Aldrich Corp. (St. Louis, MO). To generate human–mouse chimeric anti-podoplanin (chLpMab-23), appropriate VH and VL cDNAs of mouse LpMab-23 and CH and CL of human IgG1 were subcloned into pCAG-Ble and pCAG-Neo vectors (Wako Pure Chemical Industries, Ltd., Osaka, Japan), respectively. Antibody expression vectors were transfected into CHO-K1 or PDIS-5(41) using the Lipofectamine LTX reagent (Thermo Fisher Scientific, Inc.). Stable transfectants of CHO-K1/chLpMab-23 or PDIS-5/chLpMab-23 cells were selected by cultivating the transfectants in a medium containing 0.5 mg/mL of both geneticin and zeocin (InvivoGen, San Diego, CA). CHO-K1/chLpMab-23 and PDIS-5/chLpMab-23 cells were cultivated in CHO-S-SFM II medium (Thermo Fisher Scientific, Inc.). ChLpMab-23 was purified using Protein G-Sepharose (GE Healthcare UK Ltd, Buckinghamshire, England).
Production of point mutants
The amplified human podoplanin cDNA was subcloned into a pcDNA3 vector (Thermo Fisher Scientific, Inc.), and a FLAG epitope tag was added at the C-terminus. Amino acids in podoplanin were substituted with alanine using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA) with oligonucleotides containing the desired mutations. CHO-K1 or CHO-S cells were transfected with the plasmids using a Gene Pulser Xcell electroporation system (Bio-Rad Laboratories, Inc., Berkeley, CA). The point mutant cells of CHO-K1 were cultured in RPMI 1640 medium containing L-glutamine and 10% heat-inactivated FBS as described above. The point mutant cells of CHO-S were cultured in CHO-S-SFMII medium.
Flow cytometry
The cell lines were harvested after brief exposure to 0.25% trypsin/1 mM ethylenediaminetetraacetic acid (Nacalai Tesque, Inc.). After washing with 0.1% bovine serum albumin in phosphate-buffered saline (PBS), the cells were treated with primary mAbs for 30 min at 4°C, followed by treatment with fluorescein isothiocyanate (FITC)-conjugated goat antihuman or antimouse IgG (Thermo Fisher Scientific, Inc.). Fluorescence data were acquired using Cell Analyzer EC800 (Sony Corp., Tokyo, Japan).
Determination of the binding affinity using flow cytometry
LN319 (2 × 105 cells) were resuspended with 100 μL of serially diluted chLpMab-23 (0.02–50 μg/mL) or NZ-12 (0.01–20 μg/mL) followed by secondary antihuman IgG. Fluorescence data were collected using EC800. The dissociation constants (KD) were obtained by fitting the binding isotherms using the built-in one-site binding models in GraphPad PRISM 6 (GraphPad software, Inc., La Jolla, CA).
Western blot analyses
Cell lysates (10 μg) were boiled in SDS sample buffer (Nacalai Tesque, Inc.). The proteins were electrophoresed on 5%–20% polyacrylamide gels (Wako Pure Chemical Industries Ltd.) and were transferred onto a polyvinylidene difluoride membrane (Merck KGaA, Darmstadt, Germany). After blocking with 4% skim milk (Nacalai Tesque, Inc.), the membrane was incubated with chLpMab-23, NZ-12, or anti-β-actin (clone AC-15; Sigma-Aldrich Corp.) and then with peroxidase-conjugated antihuman IgG (Sigma-Aldrich Corp.) and was developed with the ImmunoStar LD Chemiluminescence Reagent (Wako Pure Chemical Industries Ltd.) using a Sayaca-Imager (DRC Co. Ltd., Tokyo, Japan).
Antibody-dependent cellular cytotoxicity
Effector cells were prepared as previously described.(7) Human peripheral blood mononuclear cells (MNCs) were obtained from leukocytes, which were separated from the peripheral blood of healthy donors. The study with human subjects was approved by the Ethics Committee of the Tokushima University. ADCC was determined using the 51Cr-release assay.(7) Target cells were incubated with 0.1 μCi of 51Cr-sodium chromate at 37°C for 1 hour. After washing three times with 10% FBS-supplemented RPMI 1640, 51Cr-labeled target cells were seeded in 96-well plates in triplicate. Human peripheral blood MNCs and chLpMab-23 or control human IgG were added to the cells. After 6 hours of incubation, 51Cr released from cells into the supernatant (100 μL) was measured using a gamma counter (PerkinElmer, Waltham, MA). The percentage of cytotoxicity was calculated using the following formula:% specific lysis = (E−S)/(M−S) × 100, where E is the release in the test sample, S is the spontaneous release, and M is the maximum release.
Antitumor activity of anti-podoplanin antibodies
CHO/podoplanin cells and HSC-2 cells were trypsinized and washed with PBS. The cell density was adjusted with PBS to 5.0 × 107 cells/mL (CHO/podoplanin) or 1.0 × 107 cells/mL (HSC-2), and 100 μL/animal of the cell suspension was subcutaneously inoculated into BALB/c nude mice. After 1 day, 100 μL of 1 mg/mL of chLpMab-23 and human IgG were injected into the peritoneal cavity of mice. Additional antibodies were injected once a week for several weeks. Human natural killer (NK) cells (5.0 × 105 cells; Takara Bio, Inc., Shiga, Japan) were injected around the tumors several times. The tumor diameter was measured every 3–7 days and was calculated using the following formula: volume = W2 × L/2, where W is the short diameter and L is the long diameter. The mice were euthanized 21 (CHO/podoplanin) or 35 days (HSC-2) after cell implantation.
Statistical analyses
All data were expressed as means ± SEMs. Student's t-test, Mann–Whitney U-test, one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparisons, and two-way ANOVA were performed as appropriate. p-Values <0.05 were considered to be significant. All statistical tests were two-sided.
Results
Epitope mapping of LpMab-23
We performed flow cytometry using several point mutants of human podoplanin to determine the epitope of LpMab-23. Seventeen podoplanin point mutants against Val50–Thr66 were transiently expressed in CHO-K1 cells. As shown in Figure 1A, LpMab-23 did not detect G54A, T55A, S56A, E57A, D58A, R59A, Y60A, or L64A. In contrast, LpMab-7,(30) a positive control mAb against human podoplanin, recognized all point mutants. These results indicate that the minimum epitope of LpMab-23 is Gly54–Leu64, which is very similar to that of LpMab-2 (Fig. 1B). Flow cytometry and western blot analyses using several glycan-deficient podoplanin cells revealed that the epitope of LpMab-23 is independent of glycans (data not shown), whereas the epitope of LpMab-2 includes O-glycan of Thr55/Ser56.(36) Here, we produced a human–mouse chimeric antibody from LpMab-23 and investigated whether chLpMab-23 possesses ADCC and antitumor activity.

Epitope mapping of LpMab-23 using point mutants of human podoplanin in flow cytometry
Production of chLpMab-23
We first checked the sensitivity of chLpMab-23 using flow cytometry. As shown in Figure 2A, chLpMab-23 reaction against human glioblastoma cell line, LN319, was observed at a very low concentration of 10 ng/mL. A positive control NZ-12, the most sensitive chimeric anti-podoplanin antibody,(44) reacted with LN319 in the same way. Both antibodies reacted with LN319 in a dose-dependent manner. We next performed a kinetic analysis of the interaction of chLpMab-23 with LN319 using flow cytometry. As shown in Figure 2B, KD of chLpMab-23 was determined to be 1.2 × 10−8 M. The KD of NZ-12 was also determined to be 1.2 × 10−8 M. NZ-12 was derived from the rat anti-podoplanin mAb, NZ-1, which possesses the highest binding affinity(8); this showed that chLpMab-23 also possesses high binding affinity against podoplanin. Western blot analysis showed that chLpMab-23 did not detect T55A and S56A (Fig. 2C), which is consistent with the results of flow cytometry using LpMab-23 (Fig. 1A). chLpMab-12,(30) a human–mouse chimeric antibody produced from another GpMab, LpMab-12 against human podoplanin, did not recognize T52A, which is consistent with our previous study.(37) chLpMab-7, a human–mouse chimeric antibody produced from LpMab-7 against human podoplanin,(45) and NZ-12 reacted with all point mutants. Because the podoplanin expression level in S56A mutant is comparatively low, the signals of chLpMab-12, chLpMab-7, and NZ-12 were lower than those of the other mutants. These results indicate that the epitope of chLpMab-23 was confirmed to include Thr55 and Ser56.
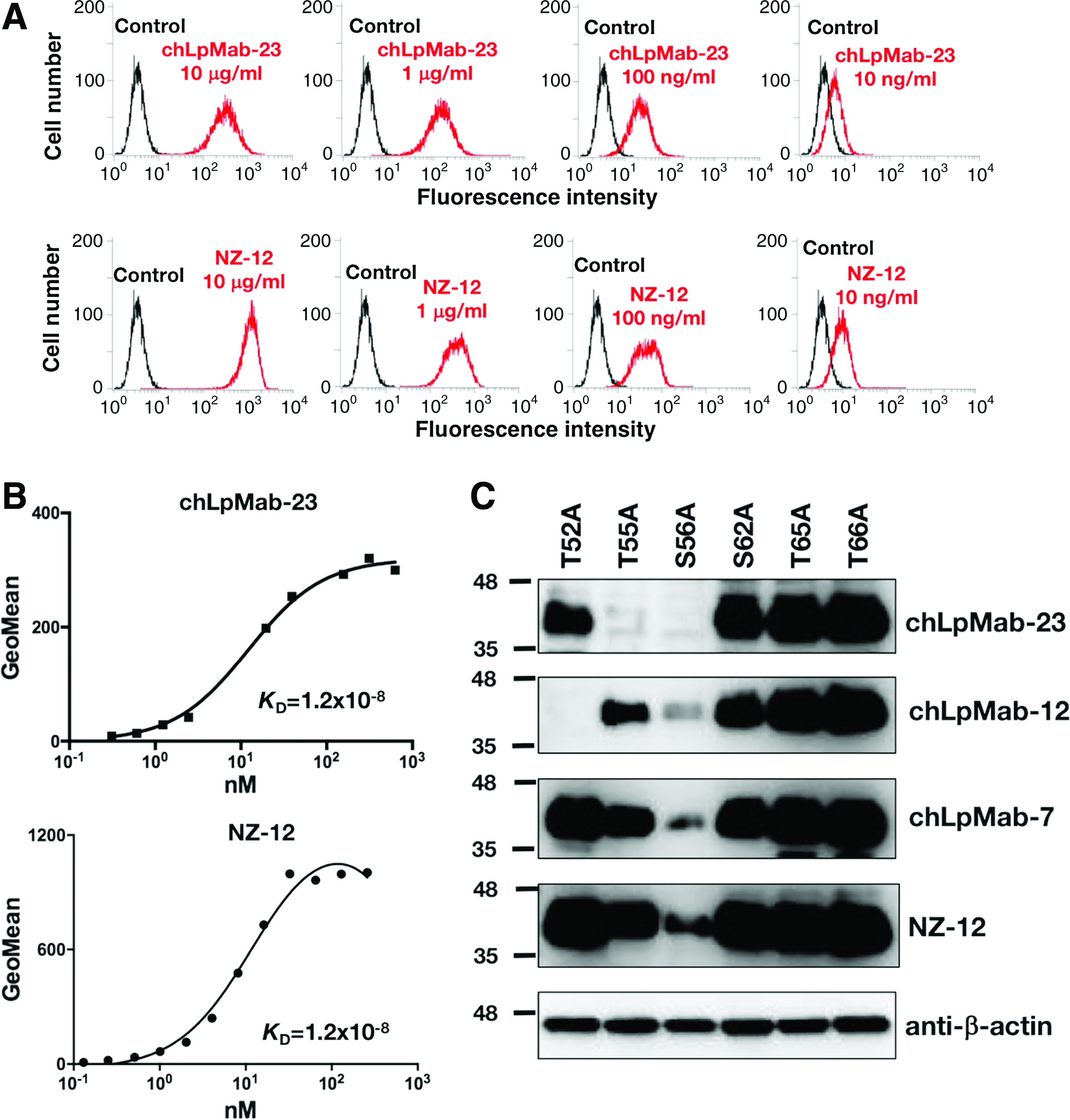
Characterization of chLpMab-23
ADCC of chLpMab-23 against podoplanin-expressing cells
Next, we investigated whether chLpMab-23 possesses ADCC activity against podoplanin-expressing cells. chLpMab-23 reacted with CHO/podoplanin in a dose-dependent manner (Fig. 3A). A positive control NZ-12 showed a similar reaction. Western blot analysis showed that chLpMab-23 specifically detected podoplanin, which is expressed in LN229 and CHO-K1 (Fig. 3B). NZ-12 also helped detect podoplanin in the same way. As shown in Figure 3C, NZ-12 showed ADCC activity against CHO/podoplanin cells. In the same way, chLpMab-23 exerted ADCC against the CHO/podoplanin cells but not against CHO-K1 cells, indicating that ADCC against CHO/podoplanin is specific against podoplanin.

ADCC and antitumor effects of chLpMab-23 against CHO/podoplanin
Antitumor activity of chLpMab-23
To study the antitumor activity of chLpMab-23 on primary tumor growth in vivo, CHO/podoplanin cells were subcutaneously implanted into the flanks of nude mice. To upregulate the ADCC activity of chLpMab-23, PDIS-5 cells (FUT-8-knockout cell line) were used for producing chLpMab-23. Core fucose-negative-type chLpMab-23, which was produced using PDIS-5, and control human IgG were injected four times into the peritoneal cavity of mice, and human NK cells were injected twice around the tumors. Tumor formation was observed in mice from the control and treated groups. However, chLpMab-23 significantly reduced tumor development compared with control human IgG on day 21 (Fig. 3D).
We further investigated whether chLpMab-23 also inhibited the tumor growth of an oral cancer cell line, HSC-2. chLpMab-23 reacted with HSC-2 in a dose-dependent manner (Fig. 4A). NZ-12 also reacted with HSC-2 in a dose-dependent manner. The reaction of NZ-12 is much more sensitive compared with that of chLpMab-23. For in vivo analysis, we used normal-type chLpMab-23, which was produced in CHO-K1 cells. HSC-2 cells were subcutaneously implanted into the flanks of nude mice. chLpMab-23 and control human IgG were injected seven times into the peritoneal cavity of mice, and human NK cells were injected three times around the tumors. As shown in Figure 4B, tumor formation was observed in mice from the control and treated groups. chLpMab-23 significantly reduced tumor development of HSC-2 compared with control human IgG on day 35 (Fig. 4C). These results indicate that administration of chLpMab-23 with the NK cells inhibited the primary tumor growth in vivo.
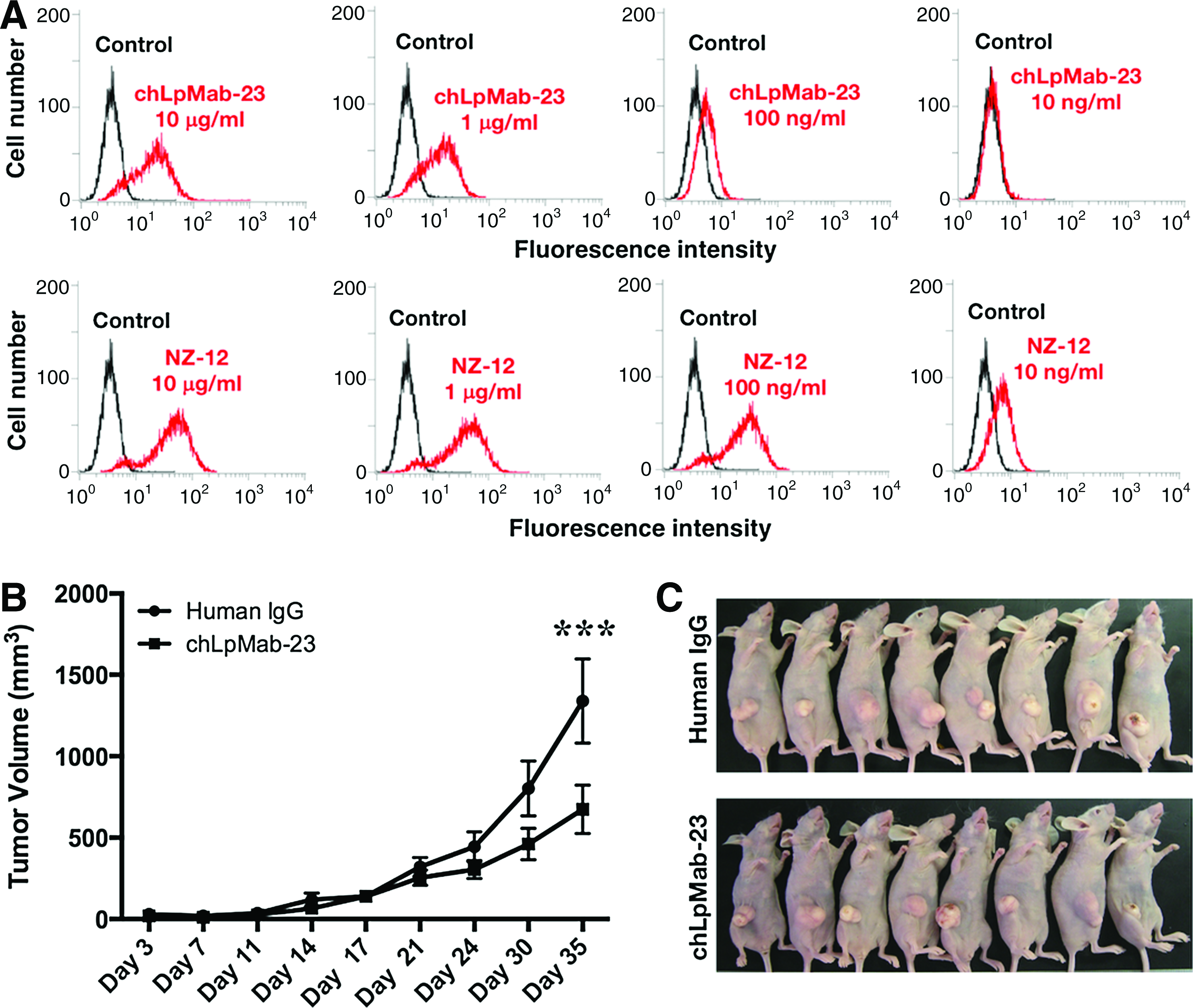
Antitumor effects of chLpMab-23 on tumor development of HSC-2 xenograft,
Discussion
In a previous study, through an immunohistochemical analysis, we showed that LpMab-23 reacted with tumor cells of human oral cancer but not with normal cells such as lymphatic endothelial cells (LECs).(42) Furthermore, flow cytometric analysis revealed that LpMab-23 reacted with podoplanin-expressing cancer cell lines but showed little reaction with normal cells including LECs and HEK-293T. Although the epitope of another CasMab, LpMab-2, was determined to be Thr55–Leu64 peptide from podoplanin, the epitope of LpMab-23 has not been clarified. In this study, we performed flow cytometry using several point mutants of human podoplanin and revealed that Gly54–Leu64 peptide from podoplanin is the LpMab-23 epitope (Fig. 1A). Furthermore, eight amino acids namely Gly54, Thr55, Ser56, Glu57, Asp58, Arg59, Tyr60, and Leu64 of podoplanin are important for LpMab-23. In contrast, seven amino acids namely Thr55, Ser56, Glu57, Asp58, Arg59, Tyr60, and Leu64 of podoplanin are important for LpMab-2,(36) indicating that epitopes of the two different CasMabs LpMab-2 and LpMab-23 are very similar (Fig. 1B).
We had not shown that LpMab-23 possesses ADCC against podoplanin-expressing cancer cells because LpMab-23 is a mouse IgG1 subclass, which does not exert ADCC. In this study, we developed a human–mouse chimeric antibody, chLpMab-23, from a mouse mAb, LpMab-23. chLpMab-23 reacted with LN319, CHO/podoplanin, and HSC-2 in a dose-dependent manner (Figs. 2A, 3A, and 4A). chLpMab-23 showed ADCC against the CHO/podoplanin cells (Fig. 3C) but not against the CHO-K1 cells, indicating that ADCC against CHO/podoplanin is specific against podoplanin. chLpMab-23 also showed ADCC against the LN319 glioblastoma cell line (data not shown). However, chLpMab-23 did not exert ADCC against malignant mesothelioma cell line NCI-H226 or lung cancer cell line PC-10 (data not shown) because podoplanin expression in those cell lines may be comparatively low. Furthermore, we investigated the complement-dependent cytotoxicity (CDC) of chLpMab-23 against several podoplanin-expressing cancer cells. However, chLpMab-23 showed no CDC against podoplanin-expressing cancer cells (data not shown). In our previous study, we showed that ADCC of anti-podoplanin mAbs is much more important for antitumor activity.(7,44) Therefore, we moved on to the investigation of antitumor activity using chLpMab-23.
To study the antitumor activity of chLpMab-23 on the primary tumor growth of CHO/podoplanin cells in vivo, core fucose-negative-type chLpMab-23 was used because core fucose-negative mAbs show higher ADCC activity.(46) chLpMab-23 significantly reduced tumor development of CHO/podoplanin on day 21 (Fig. 3D). We previously used normal-type (core fucose-positive-type) chLpMab-23 for the same xenograft model of CHO/podoplanin cells; however, chLpMab-23 did not reduce tumor development of CHO/podoplanin xenograft. We further showed that normal-type chLpMab-23 significantly reduced tumor development of HSC-2 (Fig. 4B). The different results for the CHO/podoplanin and HSC-2 xenograft models may depend on the growth speed of each xenograft or the sensitivity of human NK cells. We did not compare the antitumor activity of core fucose-negative-type and normal-type chLpMab-23 in the HSC-2 xenograft model. In the near future, we should determine the best format of chLpMab-23 for antibody therapy against podoplanin-expressing tumors.
Taken together, chLpMab-23 could be useful for antibody therapy against podoplanin-expressing cancers. The CasMabs and chLpMab-23 developed by us could be applied to novel antitumor reagents to provide strict specificity against tumor cells.
Footnotes
Acknowledgments
We thank Kimiko Takeshita for the excellent technical assistance. We also thank Junichi Takagi (Osaka University), Takeshi Murata (Chiba University), Hiroaki Uchida (The University of Tokyo), and Hideaki Tahara (The University of Tokyo) for their specialized advice. This work was supported in part by the Basic Science and Platform Technology Program for Innovative Biological Medicine from Japan Agency for Medical Research and development, AMED (Y.K.), by Project for utilizing glycans in the development of innovative drug discovery technologies from AMED (Y.K.), by the Platform for Drug Discovery, Informatics, and Structural Life Science (PDIS) from AMED (Y.K.), by JSPS KAKENHI Grant Number 26440019 (M.K.K.) and 16K10748 (Y.K.), and by the Regional Innovation Strategy Support Program from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (Y.K.). This work was performed in part under the Cooperative Research Program of Institute for Protein Research, Osaka University, CR-16-05 and CR-17-05 and by the Grant for Joint Research Project of the Institute of Medical Science, the University of Tokyo. The authors would like to thank Enago (
Author Disclosure Statement
No competing financial interests exist.