
Abstract
To design an affinity ligand for purification of antigen-binding fragment (Fab) antibody, variable domain of heavy chain antibody (VHH) phage libraries were constructed from Fab-immunized Alpaca and subjected to biopanning against Fabs. To find the specific binders, we directly applied high-throughput sequencing (HTS) analysis of the VHH sequences in the panned phages on next-generation sequencer. The efficiently enriched sequences were aligned for construction of the phylogenetic tree to be categorized into five groups. VHHs from three major groups were first selected to analyze their properties as an affinity ligand. However, those VHHs were not suitable as an affinity ligand because of lack of resistance against alkaline pH and/or difficulty in acidic elution from the affinity column. So, we further searched the candidates from minor group sequences. Among five, one VHH showed the binding ability but with low affinity against Fabs. Therefore, we improved its affinity-by-affinity maturation through error-prone PCR library techniques. The final designed VHH showed highly alkaline pH resistance and easy acidic elution together with high affinity to Fabs. These results indicate that HTS techniques combined with biopanning and followed by error-prone PCR library techniques is powerful in designing specific binders with desired properties.
Introduction
Monoclonal antibodies (mAbs) are one of the emerging biologics for human therapy in various indications. More than 70 mAbs have been approved for several diseases, including cancer and autoimmune diseases.(1–3) On the contrary, fragment antibody drugs like antigen-binding fragment (Fab) offer numerous advantages as mAb therapeutics because of their smaller size, low production cost, and easiness in protein engineering.(4–6) Recently, the keen interest has been paid toward antibody fragments (Ab-fragments) as an mAb and several mAb therapeutics have already been developed into Ab-fragments displaying alternate pharmacokinetics and other desired properties.(7,8) The production of Fab antibody fragments is expanding rapidly for the active clinical pipeline and preclinical research.(9–11) However, the purification of antibody fragments (Fabs) is a more difficult issue, because the conventional methods of immunoglobulin G (IgG) antibody using protein A or G column cannot be used because of the lack of Fc constant regions in Fabs. Several approaches have been utilized as the purification technique for Fab antibody, such as Protein-L purification method. This enables Fab purification because of the IgG kappa light chain having binding affinity to Protein-L.(12) However, for commercial purification of antibody fragment (Fab) drugs, Protein-L is somewhat limited with regard to the antibody classes, as ∼60% of mammalian IgG light chain contains kappa chain, with the remaining 40% containing lambda light chain that lack binding sites for Protein-L.(13,14) Another commonly used technique is nonaffinity-based cation exchangers.(15) However, it is very difficult to use it commercially because significant process development effort is required, which results in increasing the production cost and decreasing the production yield.(16,17) Nevertheless, the best strategy is affinity chromatography, which separates proteins based on the reversible interaction between affinity ligand conjugated to column matrix and protein interest.(18,19)
Variable domain of heavy chain antibodies (VHHs), also referred to as nanobodies, is an attractive candidate as an affinity ligand. The camelid animals, including alpaca,(20) have unique IgG antibodies in addition to the conventional IgG (IgG1) composed of two light chains and two heavy chains, heavy chain antibodies (HC Ab) composed of only heavy chain homodimers devoid of CH1 domain.(21,22) There are two types of HC Ab, classified as IgG2 and IgG3, which have short and long hinge regions, respectively, between variable antigen-binding domain VHH and Fc. This variable region (VHH) is the smallest antigen-binding single polypeptide chain naturally found in the antibody world.(23,24) VHHs are significantly more heat resistant than conventional antibodies,(25–28) and unlike the conventional whole antibodies, they can be readily produced in large amounts in bacterial expression systems.(29–31)
The phage display technology is a powerful technique to identify the functional binders. By applying this system, many target-specific antibody fragments including Fab, single-chain Fv (scFv), and others have been isolated through a biopanning selection process.(32,33) On the contrary, deep sequencing by a high-throughput sequencing (HTS) platform with a next-generation sequencer (NGS), which has been used for analysis of genomic and single nucleotide polymorphism or the antibody repertories of B cells,(34–36) has been recently applied for identification of binders from such antibody libraries in combination with biopanning.
Improving the affinity of protein–protein interaction as a therapeutic and diagnostic biomolecule is a challenging problem.(37) Several approaches have been developed for affinity maturation of antibodies derived from the original libraries, but most fall into site-directed and/or random mutagenesis.(38) Error-prone polymerase chain reaction (PCR) is one of the most popular random mutagenesis methods, where point mutations are randomly introduced into a gene.(39)
In this study, to identify the suitable VHH as a ligand for Fab purification, the VHH phage library was constructed from the immunized Alpaca with Fab and used for biopanning against Fab. We used deep sequencing techniques to find the candidates of binders. After checking and improving the properties of the candidates, we finally succeeded in design of the suitable VHH ligand for a one-step purification of Fab antibody.
Materials and Methods
Alpaca immunization
An adult male Alpaca (Vicugna pacos) was immunized subcutaneously on day 0 with 2.8 mg Trastuzumab-Fab, which was prepared by digestion of Trastuzumab (Herceptin) by papain, emulsified in Freund complete adjuvant, and on days 14 and 28 with 2.8 mg Trastuzumab-Fab emulsified in Freund incomplete adjuvant. At days 42 and 70, the same Alpaca was immunized by 1 mg Ranibizumab (Lucentis, anti-human vascular endothelial growth factor Fab fragment), and at day 56 immunized by 1 mg human constant domain of kappa light chain (CL), which was kindly donated by Dr. Yoshihisa Hagiwaka (National Institute of Advanced Industrial Science and Technology, AIST, Japan), emulsified in Freund incomplete adjuvant. Serum was collected before each injection to check the antibody response. Blood (50 mL) was collected on days 21, 63, and 84, treated with anticoagulant (0.1% ethylenediaminetetraacetic acid), and used to isolate peripheral blood mononuclear cells (PBMC) by a Ficoll-Paque method using Leucosep tubes (Greiner Bio-One). A yield of 5 × 107 cells was obtained, homogenized in RNAiso Plus (Takara Bio), and stored at −80°C until use. All genetic engineering experiments were performed under the protocol (No. 24027) approved by Gene Recombination Experiment Safety Management Committee in Kagoshima University. All animal studies were performed in accordance with Standard for Proper Conduct of Animal Experiments at Ark Resource Co., Ltd. (Kumamoto, Japan), under the approval (Protocol number: AW-130012) of the company's Institutional Animal Care and Use Committee.
Construction of alpaca VHH phage library
Total RNA was extracted from a homogenate of alpaca PBMC collected on day 84 using RNAiso Plus (Takara Bio) according to the manufacturer's protocol, cDNA was synthesized by reverse transcriptase using Oligo (dT) 20 primer from 5 mg total RNA by the SuperScript™ III First-Strand Synthesis System for Reverse Transcription-PCR (Invitrogen). VHH gene amplification was carried out by the common forward VHH-specific primer 5′-AGKTGCAGCTCGTGGAGTCNGGNGG-3′ and the reverse IgG2 short hinge-specific primer 5′-GGGGTCTTCGCTGTGGTGCG-3′ or IgG3 long hinge-specific primer 5′-TTGTGGTTTTGGTGTCTTGGG-3′, using cDNA as a template DNA. The first PCR was performed with KOD-Plus-Neo DNA polymerase (Toyobo Co.). The reaction steps were an initial denaturation (98°C for 2 minutes) and then 22 repetitions of the three-step cycle: denaturation (98°C for 30 seconds), annealing (58°C for 30 seconds), and extension (72°C for 1 minute). For the second PCR to add the restriction sites at both ends of the gene, the common forward primer harboring Sfi I site (5′-TGCTCCTCGCGGCCCAGCCGGCCATGGCTCAGGTGCAGCTCGTGGAGTCTGG-3′) and the reverse IgG2 short hinge-specific primer (5′-ATGATGATGTGCACTAGTGGGGTCTTCGCTGTGGTGCG-3′) or the reverse IgG3 long hinge-specific primer (5′-ATGATGATGTGCACTAGTTTGTGGTTTTGGTGTCTTGGG-3′) both harboring SpeI site were used. The second PCR was carried out with Gene Taq DNA polymerase (Nippon Gene Co., Ltd).
Alpaca VHH libraries were constructed using a phagemid vector pKSTV-022, which has two restriction enzyme sites, SfiI and SpeI, for cloning VHH gene at the downstream of lac promoter and pelB signal sequence, according to the method described previously.(40)
Biopanning against Fab
Two biopanning strategies, I and II, were used for enrichment of Fab-specific clones. In strategy I, 200 μg Ranibizumab/Trastuzumab-Fab was biotinylated with Lightning-Link® Rapid Biotin Conjugation Kit (Innova Biosciences). The biotinylated 20 μg Fab was reacted with the phage library (1.0 × 1011 plaque forming unit [pfu]) in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (blocking solution) for 2 hours and then added to the well of microtiter plate coated with streptavidin (SA) (500 ng in 200 μL PBS). After the 2-hour incubation, the well was washed 10 times with PBS containing 0.1% Tween-20 (PBST), 0.1 M glycine-hydrochloric acid (HCl) (pH 2.2) was added to elute the antigen-specific phage, neutralized, and then infected with Escherichia coli TG-1. After plating, the propagated bacterial cells were collected and used for the rescue of phages, according to our previous citation.(40) For the next biopanning, the phages were rescued by infection of M13 KO7 helper phages.
In strategy II, the recombinant human ErbB2/Her2-Fc protein (R&D systems) in PBS (500 ng/200 μL) was coated on a well of a 96-well microtiter plate (Nunc Thermo Fisher Scientific) and blocked with blocking solution. The phage library solution (1.0 × 1011 pfu) mixed with Trastuzumab (500 ng) in 200 μL blocking solution was added to the well coated with Her2-Fc to trap Trastuzumab complexed with phages. After 2-hour incubation, the plate was washed 10 times with PBST. The phages were eluted, neutralized, and infected with E. coli TG-1.
HTS and analysis
DNA sequences of VHH genes in the phage library were analyzed using a MiSeq system (Illumina, Inc., San Diego, CA). The MiSeq library for DNA sequencing was prepared using QIAseq 1-step Amplicon Library kit (QIAGEN) following the protocol provided by the manufacturer. The DNA sequencing sample (10 ng) was prepared by PCR using primers for both Alpaca VHH of IgG2 and IgG3, the common forward primer 5′-GGTGCAGCTCGTGGAGTCTGGGGG-3′ and the reverse primers for IgG2 5′-GGGGTCTTCGCTGTGGTGCGC 3′ and for IgG3 5′ -GTGGTTTTGGTGTCTTGGGTTC-3′, respectively. The final loading concentration was adjusted to 15 pM, following the MiSeq loading protocol. The MiSeq reagent kit v3 (Illumina) was used for long paired-end (2 × 300 bp) sequencing reactions. Run quality was monitored following the standard Illumina procedure described by the service provider. Estimation of the error rate was performed using a control DNA that was sequenced in parallel to the samples. The sequencing reads were assigned to a raw data pool based on a unique 8-bp barcode identifier and generated as a FASTQ file. The raw paired-end nucleotide sequences were merged, filtered, aligned, and trimmed by applying USEARCH Ver.8.0 (Edgar, RC) to remove low-quality and meaningless short sequences. Subsequently, an analysis program called SOPRA (Sequence Ordering Program Ranked by Amplification Factor), which is described by a custom Perl script, was used to align, cluster, and count amino acid sequence data in the Alpaca VHH phage display library.
Phylogenetic tree was constructed from VHH amino acid sequence data by the neighbor-joining method using CLC Genomics Workbench 9 (QIAGEN Bioinformatics).
Surface plasmon resonance analysis
The affinity (Kd) values were determined at 25°C using BiacoreT200 (GE Healthcare). Antigen solution (10 μM) in 40 μL of 10 mM acetate buffer (pH 4.5) was injected to research-grade CM-5 sensor chips (GE Healthcare) to immobilize the antigen by the amine coupling protocol supplied by the manufacturer. The 6.25–100 nM different concentrations of VHH solutions in HEPES buffered saline (HBS) buffer were injected for association reaction; subsequently HBS buffer was loaded for dissociation reaction. The sensor chips were regenerated by 10 mM glycine-HCl buffer (pH 2.0) containing 0.5 M sodium chloride (NaCl). The sensorgrams were analyzed using a 1:1 binding model on BIA evaluation software to determine equilibrium dissociation constants (Kd) and kinetic parameters (ka and kd).
Reconstruction of VHH antibody genes from HTS-identified sequences
Target VHH genes identified by HTS analysis were generated by PCR using complementary determining region 3 (CDR3)-specific primers for each clone. The upstream region of the VHH gene was generated through PCR by the combination of the forward primer for 5′ end region of the VHH gene and the reverse primer for CDR3-specific primer. On the contrary, the downstream region of the VHH was also generated by the combination of the CDR3-specific forward primer and the reverse primer for 3′ end region of VHH. Later, both fragments were ligated by overlap PCR, and the restriction sites for SfiI and SpeI were added to 5′ and 3′ ends of VHH gene by PCR using the paired-end primers KSB-610 (5′-TGCTCCTCGCGGCCCAGCCGGCCATGGCTCAGGTGCAGCTCGTGGAGTCTGG-3′) and KSB-344 (5′-ATGATGATGTGCACTAGTTTGTGGTTTTGGTGTCTTGGG -3′). The generated genes were digested with both restriction enzymes and ligated to phagemid vector pKSTV02 for phage generation.
Construction of VHH mutant library by random mutagenesis on error-prone PCR
For construction of mutant phage display library, error-prone PCR was carried out using GeneMorph II Random Mutagenesis Kit (Stratagene). Plasmid DNA (1 ng) of pKSTV-02 phagemid vector harboring VHH gene was used as a template DNA for error-prone PCR using a set of primers, KSB-610 and KSB-344. The mutagenic PCR reaction was performed with the following thermal conditions: Initial denaturation at 96°C for 5 minutes, then 35 cycles of denaturation step at 96°C for 30 seconds, annealing step at 60°C for 30 seconds and extension step at 72°C for 1 minute, followed by final extension at 72°C for 10 minutes. The mutated VHH gene fragments (∼400 bp) was digested with the restriction enzymes SfiI and SpeI and inserted to linearized phagemid vector pKSTV02 (1:3 molecular ratio of vector and insert DNA) by ligation reaction at 16°C for 12 hours using T4 DNA ligase (Nippon gene). The ligation product was purified by phenol–chloroform extraction method.(41) Purified DNA was transformed to competent cells of E. coli TG-1 (Lucigen Co.) by electroporation (MicroPulser electroporator; BioRad). A small part of the transformers was used for a titer check to measure the diversity of the VHH genes, and the rest were subjected to plate culture on 2TYAG medium at 30°C for 12 hours to make the library stock. For construction of VHH mutant phage library, the phages were rescued from the bacterial library stock by infection of helper phage, as described previously.(40)
Preparation and affinity chromatography on VHH-conjugated column
VHH was produced in E. coli HB2151 cells infected with cloned phages and purified on HisTrap excel columns (GE Healthcare) from the periplasmic fraction of the bacteria.(42) Purified VHH (5 mg) was immobilized on a HiTrap™ NHS-activated HP column (1 mL; GE Healthcare), and the column was blocked with 0.5 M ethanol amine 0.5 M NaCl (pH 7.5), according to the manufacturer's instructions. Ranibizumab/Trastuzumab-Fab (1 mg) with binding buffer (50 mL, 20 mM phosphate, pH 7.0) was loaded into the column at a flow rate of 1 mL/min using Profinia Purification System (Bio-Rad). After washing the column, binding Fab antibodies were eluted with 100 mM glycine-HCl, (pH 3.0). The eluate was immediately neutralized with one-tenth volume of neutralizing buffer (1 M Tris-HCl, pH 8.0) and the absorbance at 280 nm was measured to determine the yield of recovered antibody.
Results
Construction of phage libraries and enrichment of phages by biopanning
The phage libraries from short (IgG2-derived) and long hinge (IgG3-derived) VHH were independently constructed. The library sizes were 2.7 × 107 for short hinge and 5.7 × 107 for long hinge VHH. First, to get the specific binders, the conventional biopanning was examined against Fab directly coated on the plastic plate.(40) After cloning, several positive phages in enzyme-linked immunosorbent assay (ELISA) were successfully isolated. However, all the obtained clones failed to bind the soluble form of Fab on surface plasmon resonance (SPR) analysis on BIAcore (data not shown). Therefore, we changed the biopanning method to the strategies I and II (see details in Materials and Methods section). In strategy I, the phage library was mixed with biotinylated Fab (Ranibizumab or Trastuzumab-Fab) and added to SA-coated ELISA plate. In strategy II, the phage library was reacted with Trastuzumab and then the complex was trapped by HER2-Fc coated on ELISA plate. In both cases, the enrichment of the specific binders was observed in ELISA (data not shown).
NGS analysis
To identify the sequences of VHHs binding to Fab, we performed HTS analysis of the phage libraries before/after biopanning (summarized in Supplementary Tables S1 and S2). The frequencies (%) of the reads of the VHH genes in the phage libraries by NGS before and after biopanning were compared, and the changes of the frequencies of every VHH sequence through biopanning were calculated (Supplementary Table S3). This change of the frequency, named amplification factor (folds) can be considered to reflect the efficiency in increase of the number of individual VHH clones through biopanning. Therefore, it can be a good indicator to identify good binders. The top 18 VHHs showing amplification factor >20 in each round of biopanning were selected (Fig. 1a) and aligned using neighbor-joining method to construct the phylogenetic tree (Fig. 1b). The tree indicated three highly homologous groups (groups A, B, and C) showing >92% identity and sharing the common CDR3 among the members of each group. On the contrary, nonhomologous sequences can be divided into two groups: one contained Cys residue in CDR3 (group X) and the other did not (group Y).
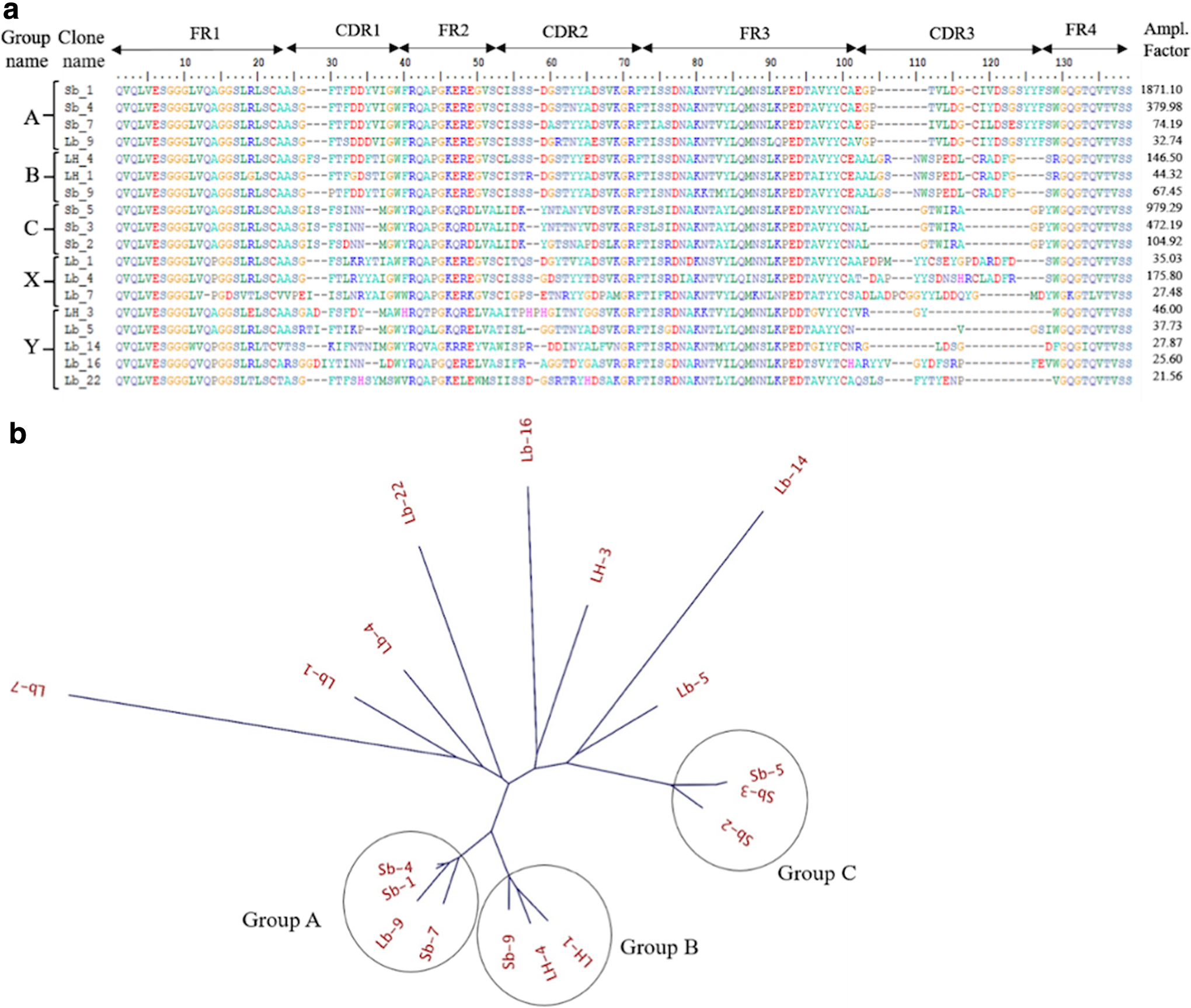
HTS analysis.
Characterizations of VHH antibodies
The representative clones from groups A (S_1), B (LH_4), and C (S_5) were prepared (Fig. 2a), and their binding properties were checked in ELISA (Fig. 2b). The VHH from each group showed the highly specific binding toward both Ranibizumab and Trastuzumab-Fab. The SPR analysis indicates that groups B and C VHHs are strong binders (Kd: <35 nM) but group A is a weak one (Kd: 200 nM), as given in Supplementary Figure S1 and Table 1.

Binding properties of groups (A, B, and C) VHH antibodies.
Summary of Binding Affinity Parameters of Typical VHH Clones from Groups A, B, and C
VHH, variable domain of heavy chain antibody.
Although the groups B and C VHHs seemed suitable for affinity ligands because of their high affinity, we found that groups A and B contained Cys residues on CDR3 region (Cys112 on group A and Cys108 in group B) (Fig. 1a), which are probably cross-linked to Cys52 in CDR2 region by disulfide bond, stabilizing the structure of CDR3. However, such disulfide bonds are easily reduced by reducing agent and broken down by alkaline pH.(43) From these considerations, groups A and B VHHs cannot be used as a ligand for affinity column that is exposed to alkaline pH in regeneration of the column.
Subsequently, we checked the acidic (pH 3.0) elution profiles of Fabs from the prepared VHH-conjugated column, as the difficult elution from the column is problematic, because it leads to time-consuming elution and reduction in recovery yield of interest protein. The column conjugated with Group C VHH, however indicated broad (difficult) elution from the column (Supplementary Fig. S2b) by acidic (pH 3.0) elution in contrast to sharp (rapid) elution in group B VHH-conjugated column (Supplementary Fig. S2a).
Binding properties of VHHs from group Y
As it was found that groups A, B, and C VHHs are not suitable for affinity ligands as described previously, we started to find other candidates. We focused on group Y, excluding group X, because group X also contained Cys residues on CDR3, as did groups A and B. Five genes of VHHs (LH_3, Lb_5, Lb_14, Lb_16, Lb_22) in group Y were reconstructed by PCR and used for the preparation of phages. The prepared phages were tested in binding to Fabs in Figure 3a and LH3 phage only showed the binding to Fabs. Therefore, LH3 protein was prepared (Fig. 3b) and further subjected to binding analysis on BIAcore (Fig. 3c). The results indicate a clear binding of LH3 to Fabs, but with low affinity (Kd: 187 nM).

Binding analysis of group Y VHHs.
Affinity improvement of Fab-specific VHH antibodies
We tried to improve the affinity of LH3 VHH by using mutational phage library constructed by error-prone PCR. The constructed library contained 1.4 × 108 unique members. After four rounds of biopanning against Fabs under the strict conditions, the phages were cloned to analyze Fab binding in ELISA. Seven clones harboring a single-point mutation and showing higher binding as compared with the parent clone (LH3) were selected (Fig. 4) and subjected to affinity measurement. The affinity parameters of these mutants are summarized in Table 2. Among them, three mutants (LH3-S30N, -Q39E, and -H56Y) showed 1.5- to 2.5-fold higher binding affinities than parent LH3, indicating these mutations can contribute to increase the affinity (Fig. 5). Therefore, we combined these mutations to make stronger binders.

Amino acid sequences of VHHs with single-point mutation obtained from error-prone PCR library. The different colors of the characters in the sequences reflect the properties of amino acids.
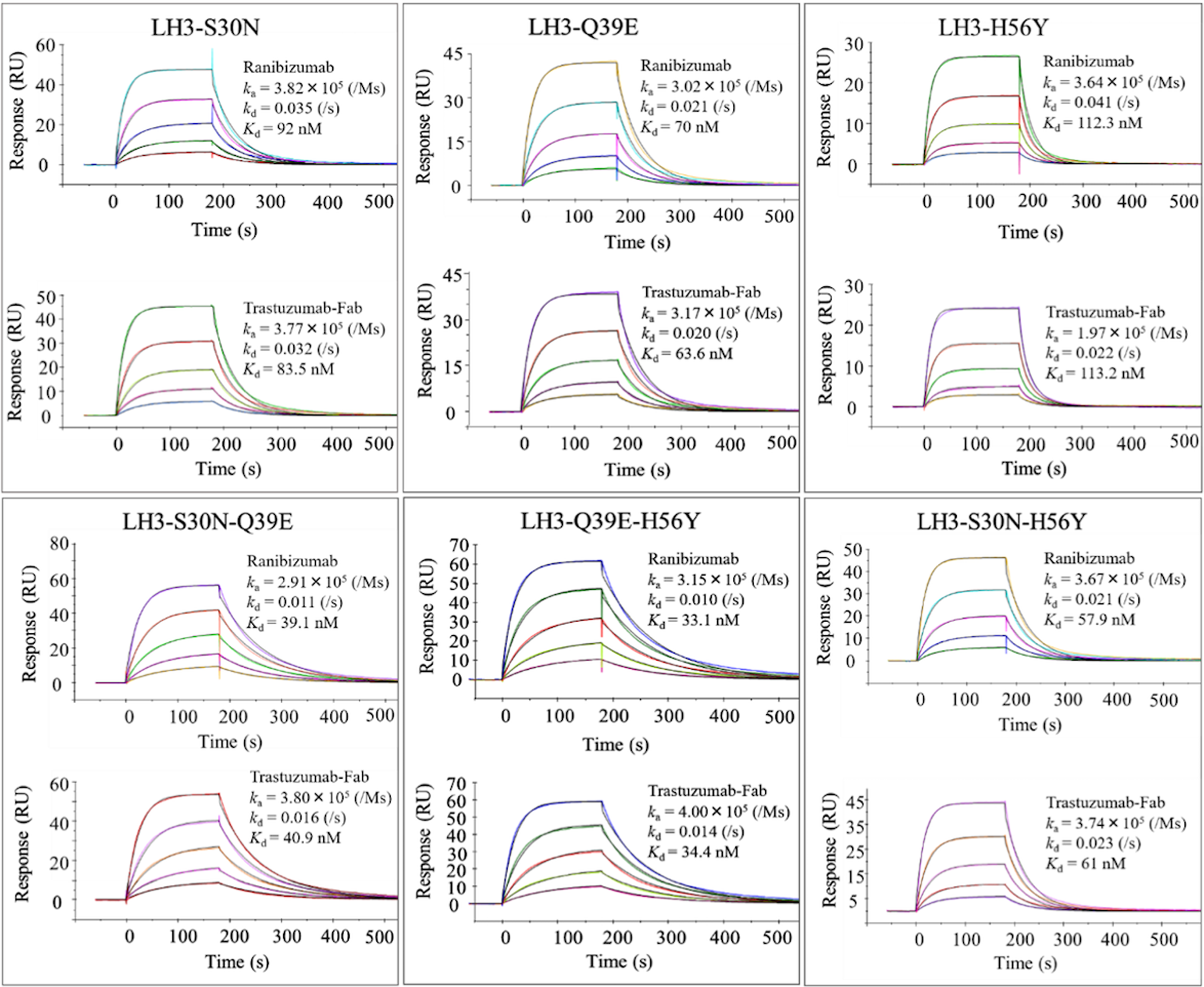
Representative SPR sensorgram binding profiles for affinity maturation. Single point mutants LH3-S30N, Q39E, and H56Y and the lead double mutants LH3-S30N-Q39E, Q39E-H56Y, and S30N-H56Y. The 6.25–100 nM different concentrations of VHH range used in each experiment. The sensorgrams were analyzed using a 1:1 binding model on BIA evaluation software to determine equilibrium dissociation constants (Kd) and kinetic parameters (ka and kd). Each mutant VHH has two sensorgrams surrounded in a box, upper panel for kinetic analysis of VHH antibody to Ranibizumab and lower panel to Trastuzumab-Fab by SPR on BIAcore-T200.
Summary of Binding Affinity Parameters and Differences in Binding Free Energies (ΔΔG) of Single-Point, Double and Triple Mutant VHHs Derived from the Parent LH3 Clone
Differences of experimental values in binding free energies (ΔΔG, kcal/mol) and theoretical values in parentheses.
Double and triple mutations at the numbers 30, 39, and 56 were combined and introduced onto the LH3 sequence. Three double and one triple mutant VHHs (Supplementary Fig. S3a) were subjected to ELISA to confirm the binding specificity to Fabs (Supplementary Fig. S3b). SPR analysis (Fig. 5) indicated that the double mutants clearly enhanced their affinity three to six-fold. The triple mutant (LH3-S30N-Q39E-H56Y) showed 10-fold higher affinity (Kd: 18 nM) than the parent VHH (Fig. 6a), demonstrating the additivity effects on these mutations in affinity. The binding free energies of the mutants are summarized in Table 2.
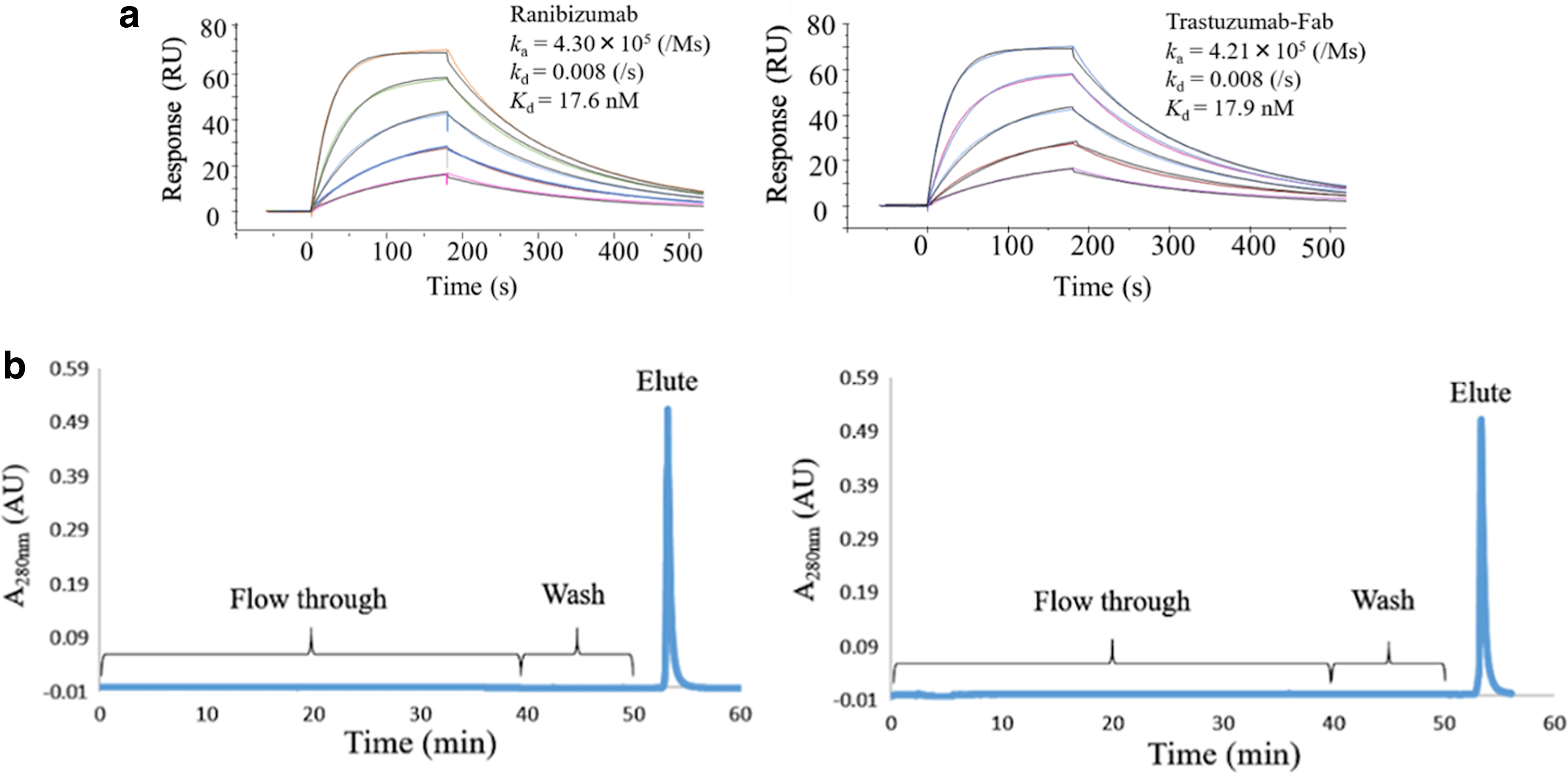
Binding and elution profile analysis of triple mutant VHH.
Affinity chromatography by VHH-conjugated column to purify Fab
To check the usability of triple mutant as affinity ligand for the column chromatography, we first tested the acidic elution profile. To the column (1 mL) conjugated with the triple mutant VHH (5 mg), 50 mL of Fab (l mg) solution was applied. After washing the column, the Fab was eluted with 100 mM glycine-HCl (pH 3.0). As given in Figure 6b, the elution was rapidly carried out, demonstrating the suitable profile as an affinity ligand.
Subsequently, the resistance of this triple mutant against alkaline pH was checked. The column was exposed to 0.1 M sodium hydroxide solution for 15 and 30 minutes at room temperature. After this regeneration process, the binding capacities of the alkaline-treated column were examined. The changes of the capacity to absorb Fabs are indicated (Supplementary Fig. S4). The engineered triple mutant VHH maintained its capacity up to 87% even after 30-minute treatment, but group B VHH (containing Cys residue on CDR3) rapidly lost its binding capacity to ∼50%, indicating the advantages of triple mutant VHH in alkaline resistance.
Discussion
When we tried to enrich the specific phages from the immunized phage library by biopanning, we first used the plastic plate physically coated with Fabs.(40) However, the VHH clones obtained by that method did not show binding ability to Fabs immobilized on the chip of BIAcore through amine coupling.(44) It was considered that probably because the structure of Fab molecule was changed to be unnatural through the process of its absorption into the surface of plate, VHHs recognizing the neoepitopes newly generated on unnatural VHH structure were enriched preferentially. Therefore, the panning method was changed into two different strategies, I and II, using biotinylated-Fab (I) and Her2-Fc coated on the plate (II). The VHHs (groups A, B, and C) showing high amplification factor on HTS analysis after biopanning showed the binding abilities to Fabs on SPR analysis. This suggests that we must select the best/better biopanning method corresponding to the required properties of VHH.
HTS analysis was proven very powerful in identifying the specific clones from the huge members of VHHs enriched by biopanning.(45) The VHH sequence (Sb_1) in group A with the highest amplification factor showed >1800-fold amplification factor, and the VHHs (LH_4 and Sb_5) from the groups B and C also showed 146- and 979-folds, respectively (Fig. 1a). These sequences showing the high amplification factor also have the high frequencies (%) in the libraries after biopanning—6.4%, 0.25%, and 0.36% for Sb_1 (group A), LH_4 (group B), and Sb_5 (group C), respectively. These high values of the frequencies mean that these major clones can be easily isolated by the conventional biopanning followed by cloning and screening. However, the LH_3 sequence that we finally selected possessed 46-fold amplification factor and only 0.07% frequency after biopanning (Supplementary Table S3), meaning the extreme difficulty in isolating such a low-frequency clone by the conventional screening. Furthermore, although we desired the VHHs with no Cys residues in CDR3,(43) to find out the VHH sequences (groups C and Y only) to satisfy this requirement would be very laborious without HTS analysis. Such a rare clone can never be found out without NGS analysis, demonstrating that NGS analysis combined with biopanning is very powerful in selection of specific binders with desired sequences and properties.
It is valuable to attain the affinity improvement of LH_3 ten-folds more (Fig. 6a and Table 2). Through biopanning from the mutational library constructed by error-prone PCR covering whole sequence of VHH, the effective mutations that improve the affinity were found on S30N in CDR1, Q39E in Frame2, and H56Y in CDR2 (Fig. 4 and Table 2), but not on CDR3. As LH_3 has short CDR3 composed of four residues (Fig. 1a), it seems to be difficult to engineer this region for affinity improvement. As a result, the target residues toward affinity improvement were not directed to CDR3 region. The contribution of individual mutation to the affinity was 0.3–0.6 kcal/mol on average in the binding energy, but double mutations combined by two mutations showed the increase of 0.6–1.0 kcal/mol and triple mutations combined by three mutations did the increase of 1.4 kcal/mol. Therefore, these mutations can contribute independently to affinity, indicating a clear additivity in the change of mutational energy (Table 2). In this way, without mutations on CDR3, through the accumulation of the small mutational energies, the increase of affinity was attained, indicating the method employed here and/or mutations found can be applicable for engineering of other VHHs.
We considered that three properties of VHH were desired as an affinity ligand. The first is strong affinity less than several tens of nanomolars at least. The second is easy acidic elution profile, which means rapid decrease in affinity at low pH as compared at neutral pH.(46) The third one is the resistance against alkaline pH.(47) The finally designed LH_3 triple mutant satisfied all these three conditions, indicating a successful design of an affinity ligand for Fab purification. On the contrary, LH_3 triple mutant recognizes Fab with kappa light chain only but not with lambda light chain (data not shown). Although we failed to isolate VHHs specific to Fab with lambda light chain in this study, we could obtain VHHs recognizing both types of Fab or Fab with lambda light chain by using the different immunization or biopanning methods in near future.
In conclusion, we successfully fabricated VHH as suitable affinity ligand from the immunized phage library using HTS followed by random mutagenesis. We were able to show that application of HTS on pooled phage libraries before/after biopanning is an effective and suitable identification technique of the target sequence in phage library. The combination of single-point mutation might be a guideline for affinity maturation without using computational predictions. Considering the center of attention, our refined VHH is expected to be a highly functional affinity ligand to establish a cost-effective, timesaving, and convenient human Fab antibody purification system with high yield and purity.
Footnotes
Acknowledgments
The authors thank Dr. Nobuo Miyazaki and Mr. Norihiko Kiyose (ARK Resource) for supporting animal experiments, and Mrs. Yoko Akazawa-Ogawa, Dr. Yoshihisa Hagihara (National Institute of Advanced Industrial Science and Technology) and Professor Tomonari Matsuda (Kyoto University) for helpful discussion.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.