
Abstract
Toxoplasma gondii is an intracellular protozoan parasite that can infect a wide range of warm-blooded animals. Humans as an intermediate host are infected by ingesting infectious oocytes or tissue cysts, or passing through the placenta in pregnant women. The aim of this study is producing monoclonal antibodies against a synthetic peptide from (surface antigen 1 [SAG1] or P30) protein of T. gondii. A synthetic peptide from SAG1 (P30) protein was conjugated to Keyhole Limpet Hemocyanin (KLH (and then used for immunization of two BALB/c mice. The produced antibody was purified by affinity chromatography and its specific interaction with the immunized peptide was then determined by enzyme-linked immunosorbent assay (ELISA). Immunoreactivity of the antibody was also tested by Western blot in T. gondii cell lysate. The results show that the produced antibody has excellent reactivity with the immunizing peptide and also detects a single band of 30 kDa, which corresponds to SAG1 protein. This antibody can be used as a tool in different applications in T. gondii research areas, including diagnosis, therapy, and infection inhibition.
Introduction
Toxoplasma gondii is an obligate intracellular protozoan parasite that belongs to Apicomplexa phylum, which can infect a wide range of warm-blooded animals.(1,2) The life cycle of this organism consists of a sexual cycle in the intestine of definitive hosts (cats) and an asexual cycle in various tissues of intermediate hosts (warm-blooded animals).(3,4) Humans as an intermediate host can be infected by ingesting the infectious oocytes and tissue cysts.(5) Because of vertical transmission potentiality of this parasite in intermediate hosts, congenital transmission can occur.(6) About one-third of the people in the world are infected by this protozoan.(7) Toxoplasmosis may cause severe disorders such as neurological and ophthalmological diseases in the developing process of fetuses and immune-deficient patients.(8,9) Moreover, due to severe side effects of available drugs and the emergence of resistance to some drugs, treatment of toxoplasmosis, especially in chronic forms, seems difficult.(10) Enzyme-linked immunosorbent assay (ELISA) surface antigen 1 (SAG1), as one of the five specific surface antigens of tachyzoite form,(11) is an immune-dominant protein,(12) which mediates attachment and invasion of this parasite to host cells.(13–15) The protective role of this antigen has been shown in several studies.(16–18) The aim of this study is to produce and characterize a monoclonal antibody against a synthetic peptide derived from the C-terminal part of the SAG1 protein as a tool for applications in T. gondii research areas, including diagnosis, therapy, and infection inhibition.
Materials and Methods
Immunogenic peptide designs from SAG1 protein and conjugated to carrier
A 19-mer peptide, KKSTAAVILTPTENHFTLK from the C-terminal part of the SAG1 protein was designed by the Center for Biological Sequence Analysis (CBS) prediction servers and peptide property calculator databases. A cysteine residue was added to the C-terminus end of the peptide to facilitate conjugation to the carrier protein. Immunograde peptide was purchased from Thermo Electron Corporation (GmbH, Ulm, Germany). The designed peptide was conjugated to Keyhole Limpet Hemocyanin (KLH) and bovine serum albumin (BSA) separately, by using maleimidobenzoyl-N-hydroxysuccinimide ester (MBS) (Sigma, St. Louis, MO), as described(19,20) with minor modifications. The peptide-KLH was used for mouse immunization and peptide-BSA for conjugation assessment and specificity testing of the antibody. The KLH acts as a carrier protein because of its high molecular weight and structure complexity. Therefore, it could induce the suitable immunoresponse against its own and its conjugated peptide. Confirmation of peptide to carrier conjugation was performed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). To check the efficacy of conjugation, 10 μg peptide-BSA ran on 10% SDS-PAGE for 1 hour with 100 mA using a mini-PROTEAN electrophoresis instrument (Bio-Rad laboratories, Philadelphia, PA); this process was followed by staining the gel with Coomassie Blue R-250 (Sigma). The change in mobility shift of conjugated BSA in comparison with nonconjugated BSA represents the efficiency of conjugation (Fig. 1)

SDS-PAGE analysis of peptide-BSA conjugate. Lane 1: BSA alone. Lane 2: peptide-BSA conjugate. BSA, bovine serum albumin; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Mouse immunization protocol
Two female BALB/c mice (8 weeks) were used for intraperitoneal immunization. Each mouse was immunized three times with 2-week intervals. The first immunization was performed using 10 μg conjugated peptide-KLH and 50 μL Freund's complete adjuvant (Sigma). For the second and third immunizations, 20 μg and 40 μg peptide-KLH were conjugated, respectively, in addition to 50 μL of Freund's incomplete adjuvant (Sigma). Twenty days after the last immunization, the mice were bled and assessed for titer of antibody by using the ELISA method. Four days before the fusion, 30 μg peptide-KLH with an equal volume of Alum adjuvant (Pick cell Laboratories, Amsterdam, Netherlands) was injected. Also, 24 hours before the fusion, 20 μg peptide-KLH without adjuvant was injected into the tail of the mice. The peptides will be digested by protease if they are injected without carrier; therefore, using a carrier-peptide conjugate is necessary at all times, even in the presence of Alum as adjuvant. Indeed, the KLH could preserve the peptide against the protease enzyme.
Bleeding and titration of specific antibody
Twenty days after the last immunization, mice were bled by a vertical incision of the tail vein. Serum ELISA test was performed as follows: wells of the ELISA plate (Nunc, Glostrup, Denmark) were coated with 100 μL of the immunizing peptide (20 μg/mL in phosphate-buffered saline [PBS]) at 37°C for 1 hour followed by overnight incubation at 4°C.(21–23) Then, the plate was washed thrice with PBS containing 0.05% Tween 20 (PBS-T); then, after 5 minutes, the process was followed by blocking with 2.5% of BSA at 37°C for 1.5 hours. The wells were then washed three times as above and the mouse sera added to the wells in twofold serial dilutions starting from 1:100. The plate was incubated at 37°C for 1.5 hours and washed again with PBS-T. At the next step, 100 μL of 1:1000 dilution of horseradish peroxidase (HRP)-conjugated sheep anti-mouse Ig (Avicenna Research Institute, Tehran, Iran) was added to the wells and incubated for 1.5 hours at 37°C. After washing, 100 μL of tetramethylbenzidine (Sigma) substrate was added to each well and the plate was then incubated at room temperature in the dark. After 15 minutes, the reaction was stopped by adding 30 μL of stop solution (0.16 M H2SO4) to each well. The optical density of the reaction was measured at 450 nm by an ELISA reader instrument (BioTek, Winooski, Vermont). Negative controls included omission of the coating layer and serum (as primary antibody) or combination of both. The mouse with the highest titer of specific antibody was selected for fusion.
Hybridoma production
Mouse myeloma SP2/0 cell line was used as a fusion partner. Cells were cultured in RPMI medium and 10% of fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). To collect mouse peritoneal macrophages, 5 mL of RPMI was injected into the peritoneal cavity of an unimmunized BALB/c mouse with subsequent aspiration and the peritoneal cells were collected under sterile conditions. The collected peritoneal fluid was washed twice with RPMI. The cells were incubated in RPMI with 20% of FBS for 24–48 hours at 37°C with 5% CO2. The spleen of the immunized mouse was removed in sterile conditions. To separate spleen cells, 10 mL of RPMI was injected into the spleen from different angles. The collected cells were washed twice with RPMI by centrifuging at 120 g for 10 minutes.
Cell fusion with polyethylene glycol
In a 50 mL sterile tube, SP2/0 cells were mixed with the spleen cells at a ratio of 1:10 (1 portion of SP2/0 and 10 portions of spleen cells). The cell mixture was washed twice with RPMI, and then, 800 μL of prewarmed (at 37°C) 50% polyethylene glycol (PEG) 1500 (Sigma) was added to the cell pellet slowly by simultaneous mixing. Immediately after adding the PEG, 20 mL of prewarmed RPMI was added to the tube to dilute the PEG. Cells were centrifuged twice in RPMI at 30 g and 22°C for 5 minutes. Selective hypoxanthine-aminopterin-thymidine medium was added to the cell pellet and the cells were seeded (2 × 106 cells/mL) into a 96-well cell culture plate. The cells were incubated at 37°C with 5% of CO2 and the cell growth and colony formations were monitored daily. For the large colonies (∼1 mm in diameter), the presence of antibody against the immunizing peptide was examined by ELISA using peptide alone and KLH-peptide, BSA-peptide, and KLH alone were used only as the coating antigen. After 2 weeks of incubation, 100 μL of supernatant from each well was separated and ELISA assay was performed as described above.
Antibody purification
Before the antibody purification, the isotype of the produced antibody in hybridoma culture supernatant was determined using the Isostrip method (Roche, Germany) according to the manufacturer's guidelines. The monoclonal antibody was purified from hybridoma culture supernatant by affinity chromatography. Briefly, the supernatant was filtered through a 45 μm filter (Nunc) and the pH adjusted to 7.5. Antibody purification was performed using an affinity column prepared by coupling the immunogenic peptide to CNBr-activated sepharose 4B (GE Health care, Uppsala, Sweden). The elution was performed using 0.1 M of glycine buffer pH 2.7. The pH of the eluted antibody was adjusted to 7.0 with 1 M of Tris buffer pH 9.0. The eluted antibody was dialyzed overnight against PBS pH 7.5. The ability of recognition of the purified antibody and its related peptide was measured by ELISA (by coating the immunogen peptide and peptide conjugates as described above) and the quality of purification was assessed by SDS-PAGE (Figs. 2 and 3).

The Isostrip image showed that the antibody produced against the protein peptide was IgG1.
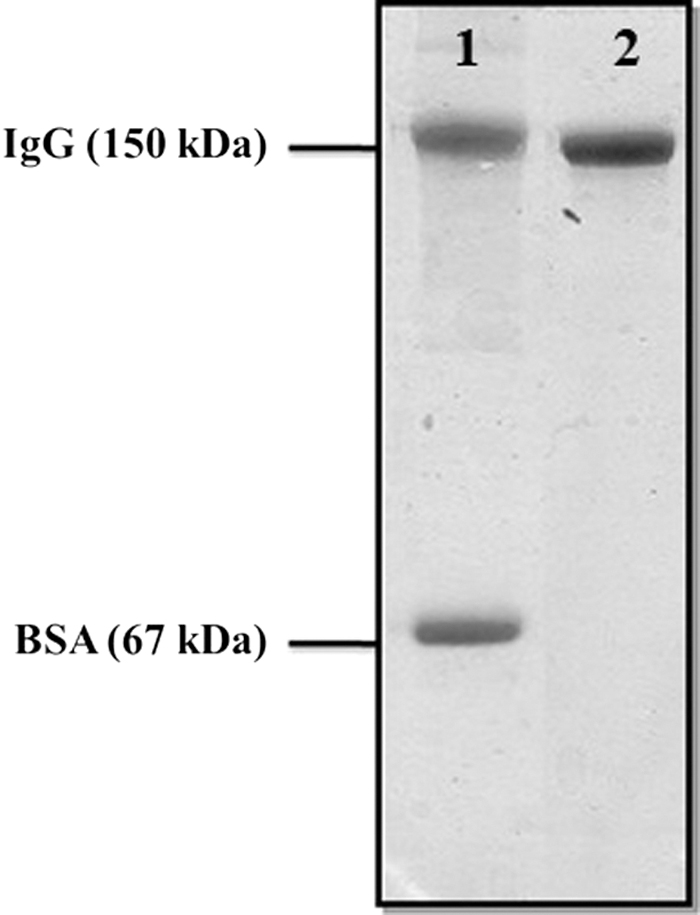
SDS-PAGE analysis of anti-SAG1 monoclonal antibody after purification. Lane 1: a mixture of mouse IgG and BSA as size marker. Lane 2: purified anti-SAG1 monoclonal antibody. SAG1, surface antigen 1.
T. gondii lysate preparation
T. gondii (resources humaines [RH] strain from Beheshti University of Medical Sciences Department of Parasitology) was propagated by intraperitoneal injection into a BALB/c mouse. After 3–5 days, the ascetic fluid was collected and centrifuged at 500 g for 5 minutes; then, the process was followed by washing the pellet thrice with PBS. Cell lysate was prepared by sonication (Hielscher ultrasonic, Germany) in 500 μL of lysis buffer containing 1% of Triton X-100, 50 mM of Tris-HCl, pH 7.4, 150 mM of NaCl, 5 mM of EDTA, 1% of protease inhibitor cocktail (Sigma), and 10% of phosphatase inhibitor (Roche Diagnostics GmbH, Penzberg, Germany) on ice. The protein concentration of lysate was measured by a bicinchoninic acid protein assay kit according to the manufacturer's instructions (Thermo Scientific, Waltham, MA).
Western blotting
Thirty micrograms of T. gondii lysate was run on 10% of SDS-PAGE with 100 V for 2 hours, and the resolved proteins were transferred to Immobilon-polyvinylidene fluoride membrane (Millipore Corporation, Billerica, MA). The membrane was blocked overnight at 4°C with 5% non-fat milk in PBS-T. All antibody incubations were performed in PBS-T containing 3% of nonfat milk. The filter was incubated with 10 μg/mL of the produced antibody for 1.5 hours at room temperature. After extensive washing with PBS-T, the membrane was incubated with HRP-conjugated sheep anti-mouse immunoglobulin (produced by Avicenna Research Institute; it is a pool of purified sheep anti-mouse immunoglobulin) for 1 hour at room temperature, followed by washing and developing with the enhanced chemiluminescence detection system (GE Health care) (Fig. 4). As a control, a lane of the membrane was incubated with the secondary antibody alone (sheep anti-mouse immunoglobulin).
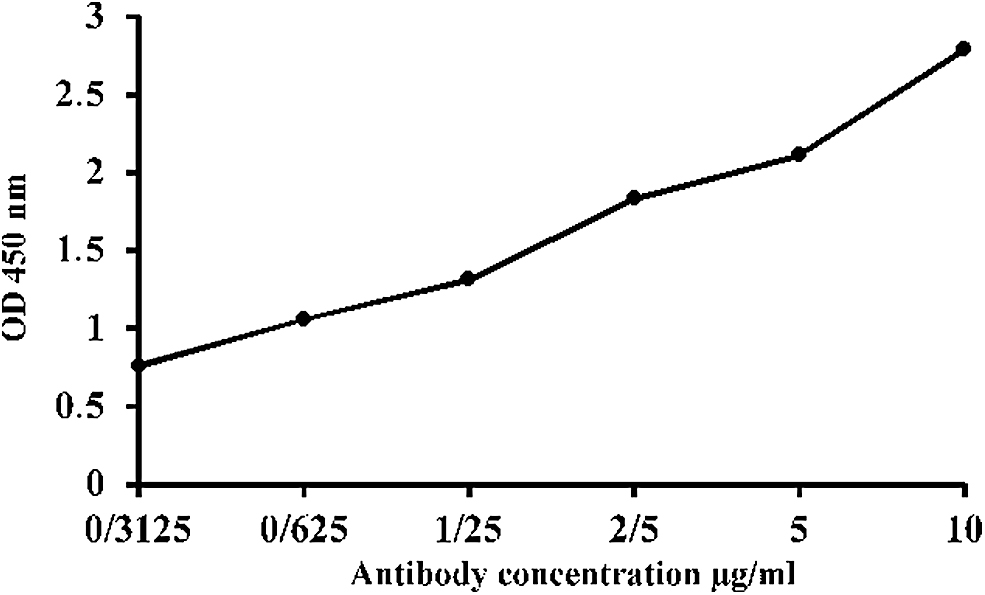
The titration chart of the purified anti-SAG1 monoclonal antibody with the immunizing peptide. This graph shows that the optical absorption rate increases with increasing antibody concentration.
Results
Peptide conjugation and antibody titration
Proper conjugation of the peptide to the carrier protein was assessed by SDS-PAGE electrophoresis. The changes in mobility shift of the peptide-BSA conjugate in SDS-PAGE gel represent the efficiency of the conjugation (Fig. 1). The presence of the antibody against the immunizing peptide in mouse sera was evaluated by ELISA (see the Materials and Methods section) before and after immunization (Fig. 4).
Evaluation of purified antibody
The Isostrip method (Roche Diagnostics GmbH) shows that the isotype of the produced antibody is IgG1 (Fig. 2). The SDS-PAGE analysis of the purified antibody revealed the presence of a single band of 150 kDa, which indicates the desired purity (Fig. 3). The purified antibody also shows excellent immune reactivity with the immunizing peptide in ELISA test, in addition to suggesting its functionality (Fig. 4).
Western blot analysis of the purified antibody
To determinate the reactivity of the purified antibody to its corresponding native protein, Western blot assay was performed. The antibody recognized a single band of 30 kDa that represents the SAG1 protein of T. gondii RH strain. As shown in Figure 5, there are some nonspecific bands in both lanes, which might be the reactivity of the secondary antibody (not our produced antibody) with unrelated T. gondii proteins or mouse immunoglobulins that have remained during the T. gondii lysate preparation.
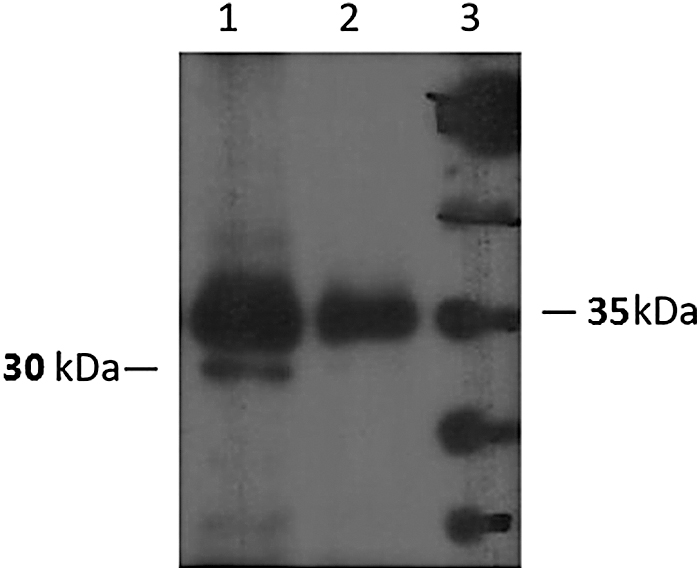
Western blot analysis of the purified anti-SAG1 monoclonal antibody. Lane 1: Toxoplasma gondii lysate incubated with both primary (the produced antibody) and secondary (HRP-conjugated sheep anti-mouse) antibodies. Lane 2: negative control (T. gondii lysate incubated with the secondary antibody conjugate alone). Lane 3: protein markers. HRP, horseradish peroxidase.
Discussion
Considering the high prevalence of T. gondii in human societies and the importance of diagnosis and treatment of toxoplasmosis in women during pregnancy, congenitally infected fetuses and newborns, and immune-deficient patients,(24–27) developing new approaches in the diagnosis and treatment of this parasite is necessary. Numerous studies have extensively investigated the SAG1 (P30) protein as a target to develop a vaccine against T. gondii by using recombinant or purified SAG1,(28–30) SAG1 DNA encoding plasmid,(31,32) and SAG1-derived peptides.(33,34) In a study conducted by Cakir-Koc in 2016, production of high titers of specific IgY antibodies against T. gondii SAG1 was performed using ELISA (Indirect, noncompetitive) and Western blot.(35) Some reports show the potential of anti-SAG1 antibodies to block the T. gondii attachment to eukaryotic cells.(36,37) In another study, Gondim et al. report a production of monoclonal antibodies against the cell wall components by using immunized mice with oocyst antigens of T. gondii.(38) In this study, we produced and characterized a monoclonal antibody against a synthetic peptide derived from SAG1 (P30) protein. In comparison to the other studies that attempt to analyze epitopes of T. gondii antigens in induction of antibody production or protection against T. gondi by using in vivo experiments,(12,39,40) we used the CBS prediction servers and peptide property calculator databases to analyze the protein structure and design a proper immunogenic peptide for antibody production. Our data might demonstrate the usefulness of in silico methods for designing antigen to produce the antibody. In 2015, Hajissa et al. were able to design a recombinant multiepitope antigen for serodiagnosis of T. gondii infection in humans, with a very high sensitivity and specificity by using antigens SAG1, GRA2, and GRA7.(41) This study also provides further evidence that the peptide can be useful as a target for antibody production against desired proteins.(42) The SAG1 protein has been proposed to be a highly protective protein in some T. gondii strains,(43) which induces high antibody levels in humans, and is diagnosed by all the serum samples from the affected patients.(44)
The antigens of tachyzoite are detectable in urine or serum samples(45); therefore, our antibody might be useful to detect the SAG1 antigen in urine samples, as a noninvasive method, to detect T. gondii infections. The antibody might also be useful for further specific antibody treatment of the infection during pregnancy when traditional treatments might not be useful to the fetus. Overall, the produced antibody in our study shows a specific interaction with the immunizing peptide as well as the SAG1 protein, which makes it a useful tool for different applications in T. gondii research areas, including diagnosis, therapy, and infection inhibition. Assays to analyze the potential of the antibody in blocking T. gondii infections might introduce novel applications for the antibody as well as the SAG1 antigens. The SAG-1 protein has a different kind of epitope than the designed peptide in this research and was a surface kind of this antigen. It also proved that we are able to design a monoclonal antibody against this peptide, which detects the SAG-1 protein with high affinity. In other words, by detecting a partial of SAG-1 protein, we could detect the whole antigen by using this produced monoclonal antibody.
Conclusion
The results of this study show that this antibody might be used as a diagnostic and research tool, where the detection or purification of T. gondii or SAG1 antigen is required. The produced antibody could also be employed for evaluation of its potential as a therapeutic tool against T. gondii using in vitro and in vivo experiments.
Footnotes
Acknowledgments
This article is equated from a Common
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by a grant from Avicenna Research Institute (ARI).