
Abstract
DNAM-1 is an activating immunoreceptor expressed on hematopoietic cells, including both CD4+ and CD8+ T cells, natural killer cells, and platelets. Since DNAM-1 is involved in the pathogenesis of various inflammatory diseases and cancers in humans as well as mouse models, it is a potential target for immunotherapy for these diseases. In this study, we generated a humanized neutralizing antihuman DNAM-1 monoclonal antibody (mAb), named TNAX101A, which contains an engineered Fc portion of human IgG1 to reduce Fc-mediated effector functions. We show that TNAX101A efficiently interfered the binding of DNAM-1 to its ligand CD155 and showed unique functions; it decreased production of the inflammatory cytokines such as interferon-gamma, tumor necrosis factor alpha, interleukin (IL)-6, IL-17A, and IL-17F by anti-CD3 antibody-stimulated or alloantigen-stimulated T cells and increased FOXP3 expression in anti-CD3-stimulated regulatory T (Treg) cells. These dual functions of TNAX101A may be advantageous for the treatment of T cell-mediated inflammatory diseases through both downregulation of effector T cell function and upregulation of Treg cell function.
Introduction
DNAM-1(
We previously generated several mouse antihuman DNAM-1 mAbs.(18) Among these mAbs, TX94 clone is a unique mAb; it most efficiently interfered the binding of DNAM-1 to CD155. Furthermore, TX94 inhibited NK cell-mediated cytotoxicity against a tumor cell line and suppressed CD8+ T cell proliferation mediated by allogeneic mixed lymphocyte reaction (MLR).(18) These functional characteristics of TX94 mAb suggest that a humanized TX94 mAb may be useful for the clinical application to immunotherapy for inflammatory diseases, in which DNAM-1 is involved in the pathogenesis. In this study, we generated a humanized TX94 mAb, named TNAX101A, which contains an engineered Fc portion of human IgG1 to reduce Fc-mediated effector functions, and show the functional characteristics of TNAX101A.
Materials and Methods
Generation of a humanized antihuman DNAM-1 antibody
The variable regions of mouse TX94 mAb was cloned from TX94 hybridoma cells. In brief, oligo dT-primed cDNA was synthesized from the total RNA extracted from TX94 hybridoma cells using the SMARTer RACE cDNA Amplification Kit (Clontech, Mountain View, CA) following the supplier's protocol. The heavy chain variable region (VH) and light chain variable region (VL) cDNAs were amplified by polymerase chain reaction (PCR) by using the universal primar A mix for 5′ primers for VH and VL provided in the Kit and 5′-GCCAGTGGATAGACAGATGG-3′ and 5′-GATGGATACAGTTGGTGCAGC-3′ for 3′ primers for VH and VL, respectively. For generation of chimeric TX94 (ChTX94) IgG1/κ antibody, the PCR-amplified fragments were cloned into a mammalian expression vector carrying kappa constant regions and human gamma-1 with amino acid substitutions from Leu to Ala at positions 234 and 235(19) (L234A/L235A; also called AA), which is known to eliminate the effector function of IgG antibodies.(20,21)
Human VH sequences homologous to the TX94 VH frameworks were searched for within the GenBank database, and the VH sequence encoded by a cDNA was chosen as an acceptor for humanization. The complementarity determining region (CDR) sequences of TX94 VH were first transferred to the corresponding positions of the human VH sequences. Next, at framework positions 48 and 71, where the three-dimensional model of the TX94 variable regions indicated significant contact with the CDRs, amino acid residues of the human VH were replaced by the corresponding residues of mouse TX94 VH. Based on the homology search with the TX94 VL framework sequences, the human Vkappa region encoded by a cDNA was chosen as an acceptor for humanization. CDR sequences of TX94 VL were first transferred to the corresponding positions of the human VL. Next, at framework position 67, where the three-dimensional model of the TX94 variable regions indicated potential significant contact with the CDRs, the amino acid residue of the human VL was replaced by the corresponding residue of mouse TX94 VL (Tyr). The VH and VL genes in ChTX94-IgG1.AA were replaced with HuTX94 VH and VL genes, respectively, and used for generation of HuTX94 (named TNAX101A).
Antibodies, reagents, proteins, and cell line
For flow cytometry analyses, antihuman CD3–V450 (UCHT1), CD4–V500 (RPA-T4), CD45RA-PE (HI100) mAbs, isotype-matched control antibodies, secondary antibodies and fluorescein-conjugated streptavidin were purchased from BD Biosciences (San Jose, CA). Antihuman FOXP3-Alexa Fluor 488 (259D/C7), CD45-Brilliant Violet510™ (HI30), CD41-PE (HIP8), and CD62P-APC (AK4) were purchased from BioLegend (San Diego, CA). For human FOXP3 staining, eBioscience™ Foxp3/Transcription factor staining buffer set (Thermo Fisher Scientific, Waltham, MA) was used. For bioassay, antihuman CD3 (HIT3a) mAb was purchased from BD Biosciences. The extracellular portion of human CD155 and CD112 fused with human IgG1 Fc portion (CD155-Fc and CD112-Fc, respectively) were generated as described.(5) BW5147 cell transfectant stably expressing human DNAM-1 was generated as described.(22)
Flow cytometry
Flow cytometric analysis was performed by using LSRFortessa flow cytometer (BD Bioscience). FlowJo software (BD) was used for data analyses.
Preparation of human peripheral blood mononuclear cell
Written informed consent was obtained from healthy donors for peripheral blood. This study was approved by the ethics committee of the University of Tsukuba, Japan. Peripheral blood mononuclear cells (PBMCs) were collected from peripheral blood by using Lymphoprep™ (STEMCELL Technologies, Vancouver, Canada) according to the manufacturer's instructions. For T cell sorting, CD4+CD8+ cells from human PBMCs were sorted by MACS system (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instruction. The purity of CD3+ cells was >95%.
Binding assay
BW5147 cell transfectant stably expressing human DNAM-1 (DNAM-1+ BW5147) was stained with ChTX94 or TNAX101A mAb, followed by PE-conjugated F(ab’)2 antihuman IgG Fc (Beckman Coulter, Brea, CA) and analyzed by flow cytometry. For competitive binding assay, human T cells from PBMCs were stained with a saturated dose of biotinylated TX94, washed with the staining medium (phosphate-buffered saline [PBS] containing 2% fetal bovine serum), incubated with TX94, ChTX94, or TNAX101A, and then stained with PE-conjugated streptavidin and analyzed by flow cytometry.
Blocking assay
DNAM-1+ BW5147 was incubated with a saturated dose of biotinylated human CD155-Fc or CD112-Fc, washed with the washing buffer, incubated with ChTX94 or TNAX101A and then stained with PE-conjugated streptavidin, and analyzed by flow cytometry. To confirm stability of the mAbs, ChTX94 and TNAX101A were incubated in PBS (pH8) for 4 weeks at 37°C or in 1 mM H2O2 in PBS for 1 week at room temperature (RT), and then the blocking assay was performed as described earlier.
Cytokine assay
PBMCs (1 × 105 cells) were incubated with TNAX101A (50 μg/mL) for 30 minutes and stimulated with antihuman CD3 mAb (10 μg/mL) in a 96-well round plate for 1, 2 or 3 days. Cytokine concentrations of interferon-gamma (IFN-γ), tumor necrosis factor (TNF), interleukin (IL)-6, IL-17A, IL-17F, IL-4, IL-10 were analyzed by Cytometric Beads Array (CBA; BD Bioscience) according to the manufacturer's instructions. FCAP Array™ software (BD) was used for data analysis.
Mixed lymphocyte reaction
PBMCs freshly isolated from healthy donor, as a responder, were incubated with TNAX101A (50 μg/mL) for 30 minutes. PBMCs from a different healthy donor, as a stimulator, were pretreated with mitomycin C (50 μg/mL). Responder cells (2 × 105 cells) and stimulator cells (5 × 105 cells) were cocultured in 96-well round plate for 2 days. Cytokine concentrations of the supernatant were analyzed by CBA as described earlier.
FOXP3 expression
PBMCs (1 × 105) freshly isolated from healthy donors were incubated with TNAX101A (50 μg/mL) for 30 minutes and cultured in the presence of anti-CD3 mAb (3 μg/mL) for 2 days. Cells were analyzed for FOXP3 expression by flow cytometry.
Platelet activation assay
Whole blood was drawn from healthy donors in the presence of citrate. Platelet-rich plasma was isolated by Optiprep™ (Abbott Diagnostics Technologies AS, Oslo, Norway) and incubated in the presence of 10 μg/mL mIgG1, TX94, antihuman DNAM-1 mAb (clone DX11 [mouse IgG1]; BD Bioscience), F(ab’)2 fragments of TX94 [TX94 F(ab’)2] or TNAX101A for 10 minutes at RT, fixed with 1% paraformaldehyde for 2 hours at 4°C. Blood samples were then stained with PE-conjugated antihuman CD41 mAb and APC-conjugated antihuman CD62P mAb and analyzed for platelet activation by flow cytometry.
Statistical analysis
GraphPad Prism 8 (GraphPad Software, San Diego, CA) was used for unpaired Student's t-test, one-way analysis of variance (ANOVA), two-way ANOVA, Smirnov–Grubbs test, and paired Student's t-test.
Results and Discussion
Generation and characterization of humanized anti-DNAM-1 antibody
For the purpose of clinical usage of an anti-DNAM-1 mAb, we first generated a chimeric TX94 mAb (ChTX94), which consists of the VH and VL regions of TX94 mAb fused with the Fc portion of human IgG1 with amino acid substitutions (L234A/L235A) for elimination of binding ability to the Fcγ receptors (Fig. 1A). Then we searched for human VH and VL sequences homologous to the TX94 VH frameworks in the GenBank database. The humanized TX94 (named TNAX101A) was generated by insertion of the CDR1, CDR2, and CDR3 regions of VH and VL of TX94 into the human VH and VL sequences selected from the database, respectively, followed by fusion of these VH and VL regions to the Fc portion of ChTX94 (Fig. 1A).
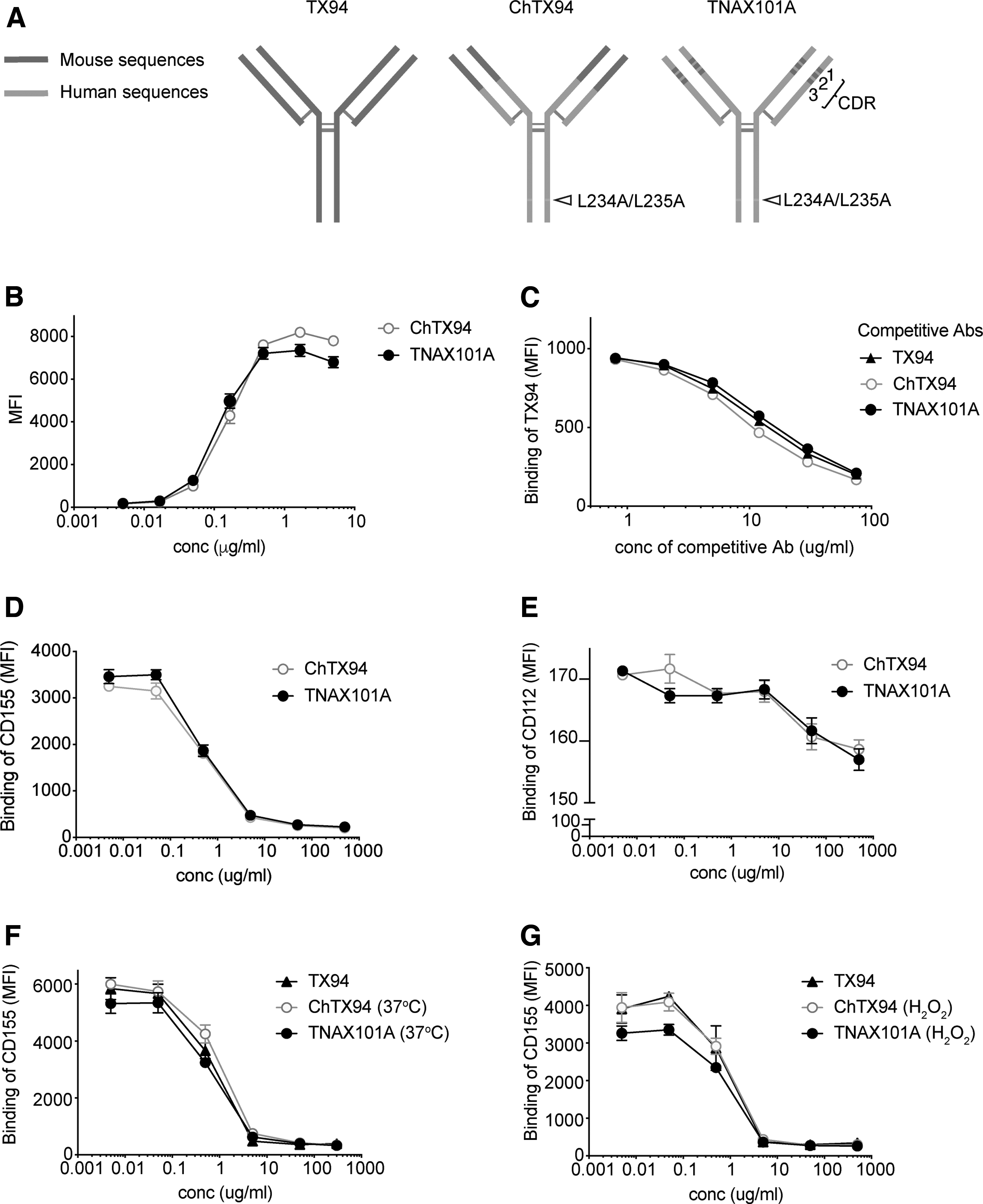
Generation and characterization of a humanized antihuman DNAM-1 antibody.
To analyze the binding ability of TNAX101A or ChTX94 to DNAM-1, BW5147 transfectant stably expressing human DNAM-1 was stained with either mAbs and analyzed by flow cytometry. As a result, the binding to the transfectant was comparable between both mAbs in a dose-dependent manner (Fig. 1B). Moreover, competitive binding assays showed that TNAX101A as well as ChTX94 and TX94 similarly replaced TX94 that had bound to DNAM-1 expressed on human T cells (Fig. 1C), suggesting that the binding affinity were comparable with each other. Furthermore, these mAbs similarly inhibited the binding of CD155-Fc and CD112-Fc to the BW5147 transfectant in a dose-dependent manner (Fig. 1D, E). TNAX101A also inhibited the binding of DNAM-1 to CD155-Fc even after it was incubated in PBS (pH8) at 37° for 4 weeks or at RT in the presence of 1 mM H2O2 for 1 week (Fig. 1F, G). Together, these results suggest that the conformation of the CDR1, 2 and 3 of TNAX101A was not changed from those of TX94 and ChTX94 and that the TNAX101A was physically and functionally stable during those conditions.
TNAX101A inhibits inflammatory cytokines production
DNAM-1 is a costimulatory molecule in T cells to promote proliferation and cytokine production.(16,23) We next tested whether TNAX101A inhibits inflammatory cytokine production by T cells. PBMCs were stimulated with anti-CD3 mAb in the presence or absence of TNAX101A. Cytokine bead array analysis of the culture supernatants demonstrated that IFN-γ, TNF-α, IL-6, IL-17A, and IL-17F concentrations were significantly lower in the presence of TNAX101A than those in the presence of control mAb at certain time points after stimulation (Fig. 2A). It seems that the appropriate time window for each cytokine measurement to analyses of TNAX101A effect may be different from each other after stimulation. In contrast, we did not observe statistically significant difference in the concentrations of Th2 cytokines such as IL-4 and IL-10. Moreover, IFN-γ and TNF-α concentrations were significantly reduced when TNAX101A was added into an MLR assay (Fig. 2B). These results indicated that TNAX101A efficiently inhibits inflammatory cytokine production by activated T cells.

TNAX101A inhibits inflammatory cytokines production. Human PBMCs from healthy donors were stimulated with anti-CD3 mAb for indicated days
TNAX101A upregulates FOXP3 expression
Human CD4+ T cells are divided into five fractions according to the expression of CD45RA and the master regulator of Treg cells FOXP3.(24) However, the expression profile of DNAM-1 on human CD4+ T cell subpopulations remains unclear. We found that although DNAM-1 was broadly expressed in each fraction of CD4+ T cells, the fraction IV, which is CD45RA+FOXP3- effector/memory conventional T (Tconv) cells, showed the highest expression of DNAM-1 among the five fractions. DNAM-1 was also expressed on FOXP3+ Treg cells, including the fractions I (naive/resting Treg cells), II (activated/effector Treg cells), and III (nonsuppressive Treg cells). We found that DNAM-1 expression was higher in the fractions II and III compared within the fraction I (Fig. 3A, B), suggesting that DNAM-1 might be involved in Treg cell activation and function.

TNAX101A upregulates FOXP3 expression.
Several evidences demonstrated that FOXP3 expression is downregulated in activated Treg cells in inflammatory conditions.(25,26) To analyze the role of DNAM-1 in Treg cell function, PBMCs were stimulated with anti-CD3 mAb in the presence or absence of TNAX101A and analyzed for FOXP3 expression by flow cytometry. We observed that FOXP3 expression was higher in anti-CD3-stimulated PBMC in the presence of TNAX101A compared within the absence of TNAX101A (Fig. 3C). These results suggest that TNAX101A may have a function to increase FOXP3 expression in activated Treg cells. Our results were consistent with the previous reports demonstrating that deficiency of DNAM-1 or DNAM-1-mediated signaling enhances Treg cell function in mouse models of acute GVHD, unilateral ureteral obstruction, and allogeneic skin transplantation.(17,27,28) Future studies were required for clarification of molecular basis of the role of DNAM-1 in Treg cell function.
TNAX101A does not activate platelets
Human platelets express FcγRIIA (CD32a), a low-affinity receptor for the constant region of IgG, which recognizes IgG immune complexes (ICs) and IgG-opsonized cells with high avidity.(29) Engagement of this receptor on platelets by ICs triggers intracellular signaling events through immunoreceptor tyrosine-based activating motif in the cytoplasmic region, leading to platelet activation and aggregation.(30) Since platelet express DNAM-1, an anti-DNAM-1 IgG antibody is likely to co-ligate DNAM-1 with FcγRIIA and induces platelet activation. To examine this possibility, human platelets were incubated with anti-DNAM-1 mAbs, including TNAX101A, TX94, F(ab’)2 fragment of TX94 and DX11 clone, and then stained with PE-conjugated anti-CD41 mAb and APC-conjugated anti-CD62P, and analyzed by flow cytometry. We found that TX94 and DX11 upregulated CD62P expression (Fig. 4A, B), indicating that these anti-DNAM-1 mAbs activated platelets. In contrast, CD62P expression was not changed after treatment with TNAX101A or F(ab’)2 fragments of TX94 (Fig. 4A, B). These results suggest that the binding ability of the Fc portion of TX94 and DX11 to FcγRIIA is critical for platelet activation, and TNAX101A does not activate platelets.
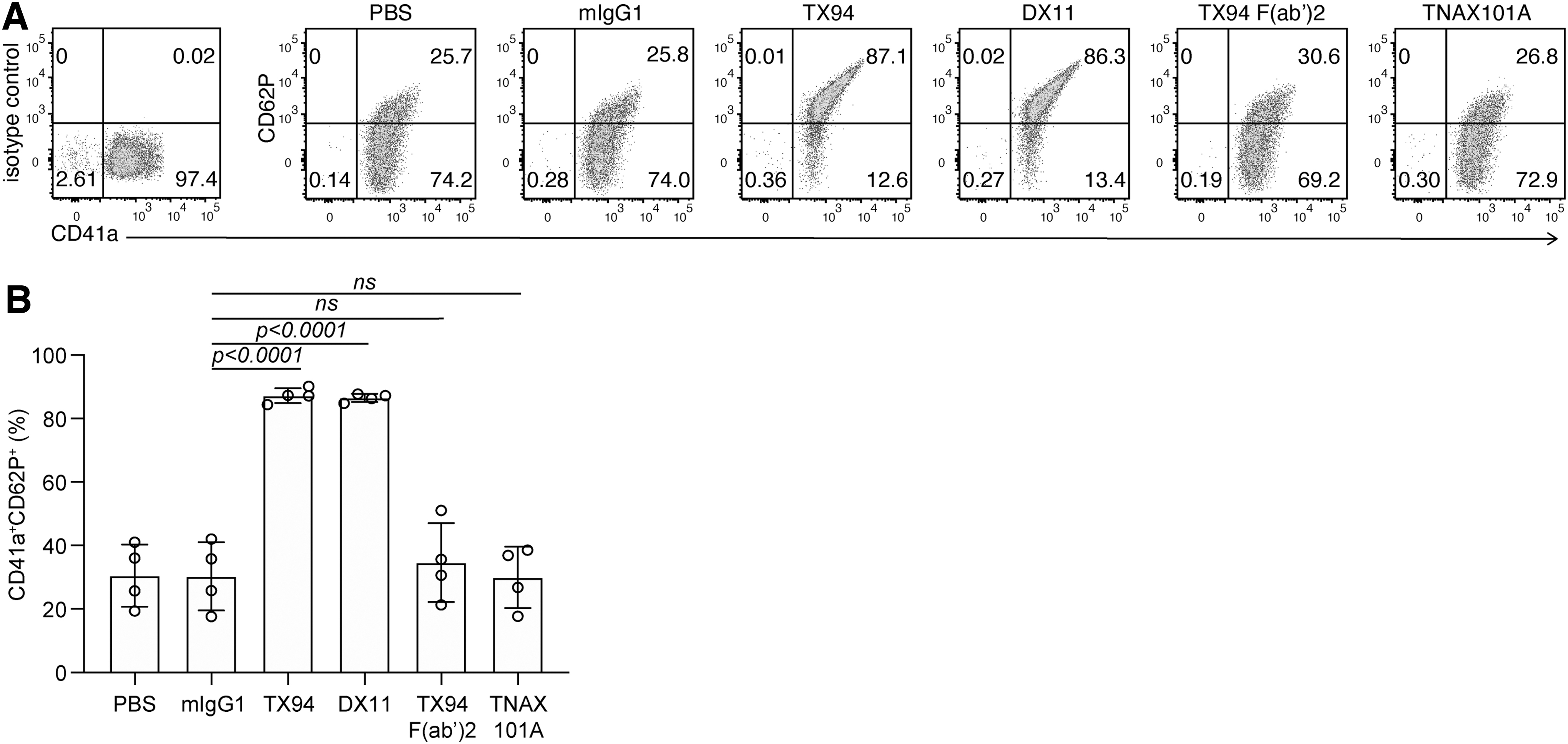
TNAX101A does not activate platelets.
Conclusion
In this study, we generated TNAX101A mAb, an Fc-engineered humanized anti-DNAM-1 mAb. TNAX101A efficiently interfered the binding of DNAM-1 to CD155 and showed unique functions; it decreased production of the inflammatory cytokines such as IFN-γ, TNF-α, IL-6, IL-17A, and IL-17F by T cells and increased FOXP3 expression in anti-CD3-stimulated Treg cells. These dual functions of TNAX101A may be advantageous for the treatment of T cell-mediated inflammatory diseases through both downregulation of effector T cell function and upregulation of Treg cell function. Of note, although TX94 activated platelets, TNAX101A showed no such an effect, suggesting that TNAX101A would not show platelet aggregation and depletion in the clinical setting. Further studies in the preclinical and clinical settings will be required to apply this mAb for the therapy of human inflammatory diseases.
Footnotes
Authors' Contributions
Y.Y.-K. performed experiments and wrote the article. K.O. and F.A. performed experiments. K.S. designed the research and supervised the project. A.S. designed the research, supervised the project, and wrote the article.
Author Disclosure Statement
Y.Y.-K. and F.A. are employees of TNAX Biopharma Corporation. K.S. and A.S. own a stake in TNAX Biopharma Corporation.
Funding Information
This study was supported by grants from the Japan Society for the Promotion of Science (JSPS KAKENHI; grant nos. 18H05022 and 16H06387 to A.S.). K.O. is a research fellow of Japan Society for the Promotion of Science.