
Abstract
Hirame novirhabdovirus (HIRRV) is a significant viral pathogen of Japanese flounder (Paralichthys olivaceus). In this study, seven monoclonal antibodies (mAbs) against HIRRV (isolate CA-9703) were produced and characterized. Three mAbs (1B3, 5G6, and 36D3) were able to recognize nucleoprotein (N) (42 kDa) and four mAbs (11-2D9, 15-1G9, 17F11, and 24-1C6) recognized matrix (M) protein (24 kDa) of HIRRV. Western blot, Enzyme-linked immunosorbent assay, and indirect fluorescent antibody technique (IFAT) results indicated that the developed mAbs were specific to HIRRV without any cross-reactivity against other different fish viruses and epithelioma papulosum cyprini cells. All the mAbs comprised IgG1 heavy chain and κ light chain except 5G6, which has a heavy chain of IgG2a class. These mAbs can be very useful in development of immunodiagnosis of HIRRV infection.
Introduction
Hirame novirhabdovirus (HIRRV), formerly named hirame rhabdovirus, is a significant viral pathogen of cultured Japanese flounder (olive flounder, Paralichthys olivaceus) where it causes high mortality.1,2 The virus has ∼11 kb antisense-single-stranded RNA genome and was classified under the Novirhabdovirus genus of Rhabdoviridae family.1,3–5 The virus can be genetically and serologically distinguished from other fish rhabdoviruses.1,4,6 Consistent with other Novirhabdoviruses, HIRRV also possesses six viral proteins, including nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), nonvirion (NV) protein, and polymerase (L).4,6,7 The apparent clinical manifestations of HIRRV infection in Japanese flounder are gonadal congestion, fin and muscular hemorrhages, and ascites. 1
Initial isolations of HIRRV were recorded in 1984 from cultured Japanese flounder and ayu (Plecoglossus altivelis) in Japan. 1 Since then, it has been isolated from other seawater fishes, namely stone flounder (Kareius bicoloratus), black seabream (Acanthopagrus schlegeli), and spotted sea bass (Lateolabrax maculatus) in Korea and China,8–10 but also from freshwater fish (grayling, Thymallus thymallus and brown trout, Salmo trutta) in Poland. 11 The earliest record of HIRRV infection from Korea was found to be in 1997 from farmed olive flounder. 12 Afterward, HIRRV has not been reported from olive flounder and other marine fish until 2012, even though infection with viral hemorrhagic septicemia virus (VHSV) is being reported frequently from olive flounder in winter to spring season.13,14 HIRRV infection was again found in cultured spotted sea bass in 2013 and black seabream in 2015,9,10 even though it is unclear why the virus reappeared.
Till date, the prevalent detection methods for HIRRV infection consists of cell culture isolation and molecular assays viz., reverse transcriptase (RT)-polymerase chain reaction (PCR) and TaqMan probe-based quantitative RT-PCR in conjunction with observation of clinical signs.9,11,14,15 Early diagnosis and appropriate implementation of prevention strategies are the ideal ways to manage HIRRV infection. Antibody-based rapid immunological assays can be an option due to its high sensitivity, low cost, and speediness.16–18 Therefore, anti-HIRRV antibody-based immunological methods could assist to enhance as well as to curtail the time required for the detection of HIRRV. However, immunological methods are not in use primarily due to lack of a robust antibody against HIRRV.
Currently, there is only one report of specific anti-HIRRV monoclonal antibody (mAb) to use. 19 Even though there is mAbs against HIRRV, 19 requirement of sensitive antibodies in developing point-of-care tests and other pathogenesis studies is highly demanded. Thus, in quest of future demands and toward the goal of production of mAbs against other than M protein of the virus, the current work is undertaken to produce and functionalize a bunch of anti-HIRRV mAbs by enzyme-linked immunosorbent assay (ELISA) and western blotting, which have a potential to be used in the immunological diagnosis of HIRRV infection.
Materials and Methods
Virus culture and isolation
An isolate of HIRRV (CA-9703) originally obtained from HIRRV-infected olive flounder in Korea was used. 12 In the present study, HIRRV was cultured in epithelioma papulosum cyprini (EPC) cell line maintained at 15°C in Dulbecco's minimum essential medium (DMEM; Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco), 150 IU/mL penicillin G, and 100 μg/mL streptomycin in 75 cm2 tissue culture flask (Nunc, Denmark). Cultured HIRRV was centrifuged (12,000 g, 30 minutes, 4°C) and the supernatant was subjected for HIRRV purification according to previously communicated protocol. 6
In brief, the virus was concentrated using polyethylene glycol (PEG-6000; Sigma) (7.5% w/v) plus NaCl (2.3% w/v) mixture and purified by ultracentrifugation (28,000 g, 2 hours) through a discontinuous gradient that comprised 20%, 35%, and 50% (w/w) sucrose in phosphate-buffered saline (PBS, 0.13 M NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.4 mM KH2PO4) buffer. The virus band was collected and sedimented by ultracentrifugation (28,000 g, 2 hours). The resultant pellet was dissolved in PBS and stocked at −80°C until used.
Production and purification of mAb
mAbs were produced by hybridoma technology as described earlier. 20 In brief, 6 weeks old five BALB/c mice were intraperitoneally injected with the purified HIRRV suspension after emulsification with Freund's complete adjuvant (Sigma) at 1:1 dilution. The mice were given booster doses with the purified HIRRV antigen by intraperitoneal injection two times at 1-week interval. The immunized spleen cells were harvested 3 days after the last immunization and combined with mouse myeloma cells (SP2/0-Ag14) at a ratio of 10:1 using PEG (MW 1500; Roche). After aminopterin and thymidine (HAT) selection in 1 × HAT (0.1 mM hypoxanthine, 4 × 10−4 mM amino protein, 2 mM thymidine in DMEM), and hybridoma colonies were screened by reacting with purified HIRRV using ELISA, and positive clones were selected by limited dilution (three times).
Animal experiments using mice were carried out in strict accordance with the recommendations of the Institutional Animal care and Use Committee of Chonnam National University (Permit Number: CNU IACUC-YS-2017-2). Isotyping of the selected clones were determined by sandwich ELISA using rapid ELISA mouse mAb isotyping kit (BD Biosciences) following the manufacturer's protocol.
Western blot analysis
The supernatants of established hybridoma were subjected to western blotting to determine the recognition site of antibodies on HIRRV. Concisely, sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 10% polyacrylamide gel was performed. 21 The gel containing purified HIRRV suspension was stained using Coomassie brilliant blue (R-250; Wako, Japan). The proteins were profiled by SDS-PAGE and then transferred onto nitrocellulose (NC) membrane as previously described. 22 The electrotransferred HIRRV proteins were reacted with produced mAbs and imaged using enzyme conjugated rabbit anti-mouse immunoglobulin G (IgG) (alkaline phosphatase; Dako) and substrate solution (0.34 mg/mL of nitroblue-tetrazolium, NBT, 0.17 mg/mL of 5-bromo-4-chloro-3-indolyl-phoshatetoluidinium, BCIP, 100 mM Tris-HCl, pH 9.5, 100 mM NaCl, 50 mM MgCl2).
The specificity of mAbs was also analyzed performing a western blot against seven types of fish virus-infected cells, viz., HIRRV (CA-9703, Kor-TY15), infectious hematopoietic necrosis virus (IHNV isolate RtUi02), and spring viremia of carp virus (SVCV)-infected EPC cells, VHSV (FYeosu05 isolate)-infected fathead minnow cells, infectious pancreatic necrosis virus (IPNV isolate VR-299), marine birnavirus (MABV isolate NC-1)-infected chinook salmon embryo-214 (CHSE-214) cells, and nervous necrosis virus (NNV isolate SGYeosu08)-infected striped snakehead (SSN-1) cells. The monolayer of respective cells was inoculated with 100 μL virus in a 24-well plate. Cells were harvested on reaching 70% cytopathic effect by decanting supernatant followed by washing the cell pellet with 0% FBS-DMEM two times. To the harvested cells, 100 μL of SDS-lysis buffer (166 mM Tris, pH 6.8, 5.3% [w/v] of SDS and 13% [v/v] of 2-mercaptoethanol) were added and subsequently boiled at 100°C for 3 minutes for the sample preparation for SDS-PAGE.
Enzyme-linked immunosorbent assay
The indirect ELISA was used to detect the specificity of mAbs against two HIRRV isolates (CA-9703 and Kor-TY15 from black seabream 9 ) and six different other viruses (VHSV FYeosu05 isolate, IHNV RtUi02 isolate, SVCV, IPNV VR-299 isolate, MABV NC-1 isolate, and NNV SGYeosu08 isolate), also to the supernatant of EPC cells and DMEM10 as previously published. 23 Concisely, these antigens were diluted 320 times with distilled water, and 50 μL each was dispensed into 96-well microplates (Greiner bio-One, Austria) and coated by incubating at 37°C for overnight. The ELISA plate wells were blocked with 5% blocking buffer (skim milk powder in PBS) at 25°C for 1 hour.
The wells were incubated with mAb solution prepared in blocking buffer for an hour at 25°C after washing thrice with PBST (PBS-0.05% Tween-20). Positively reacted anti-HIRRV mAbs were detected with horseradish peroxidase conjugated goat anti-mouse IgG (1:1000 dilution with blocking buffer; Dako) and substrate solution (1 mg/mL ο-phenylenediamine, 0.03% H2O2, 100 mM Na2HPO4, 50 mM citric acid). Absorbance value (A490) was read-out using a microplate reader (SpectraMax™ 340PC384; Molecular Devices) after stopping the reaction with 100 μL of 2 N H2SO4. The results were given as the average of OD values in duplicate and graphically illustrated.
Immunofluorescence
CHSE-214 cells were grown on 96-well microtiter plates (Nunc) maintained at 15°C in DMEM10. The cells were infected with HIRRV (CA-9703), IHNV, and VHSV at a multiplicity infection of 0.01–0.001 and incubated at 15°C for 48 hours. PBS mock-infected cells were used as the negative control. The medium was removed after incubation, and wells were rinsed with PBS followed by 10% formalin fixation at room temperature for 1 hour. After repeated three times wash with PBS, the wells were blocked with 5% skim milk for 1 hour.
The respective cells were reacted with hybridoma supernatants of anti-HIRRV mAbs at 25°C for 1 hour and anti-IHNV mAb and anti-VHSV mAbs were used as the positive controls against IHNV and VHSV, respectively. The cells were incubated for 1 hour at 25°C in the dark with goat anti-mouse IgG-FITC (1:50; Novus). Five times stringent wash with PBS was done after each reaction step. The fluorescence was observed under fluorescent inverted microscope (Nikon DIAPHOT 300, Japan).
Results and Discussion
Generation of anti-HIRRV mAbs
Several mAbs were already established and concurrently used for the detection of other fish rhabdoviruses such as VHSV and IHNV.17,24–26 There were fewer reports regarding the development of anti-HIRRV antibodies1,6 apart from one on the characterization of an anti-HIRRV mAbs against recombinant M protein. 19 We produced and characterized seven anti-HIRRV mAbs against conventionally purified viral particles for the benefit of immunodiagnosis of HIRRV infection in olive flounder.
Purification of HIRRV was done by PEG and sucrose gradient ultracentrifugation and the virus band showed up in between 20/35% gradient interface. The resultant virus band displayed five major protein bands with a molecular weight of 189, 58–63, 42, 31, and 24 kDa, respectively, in SDS-PAGE (Fig. 1A). It identified five structural proteins of HIRRV from olive flounder as L (156 kDa), G (68 kDa), N (46.4 kDa), M 1 (P, 26.4 kDa), and M 2 (M, 19.9 kDa). 6 Even though the five protein bands in the electrophoretic pattern were similar to the five major proteins of HIRRV, 6 but slight variations in the band sizes prevails.
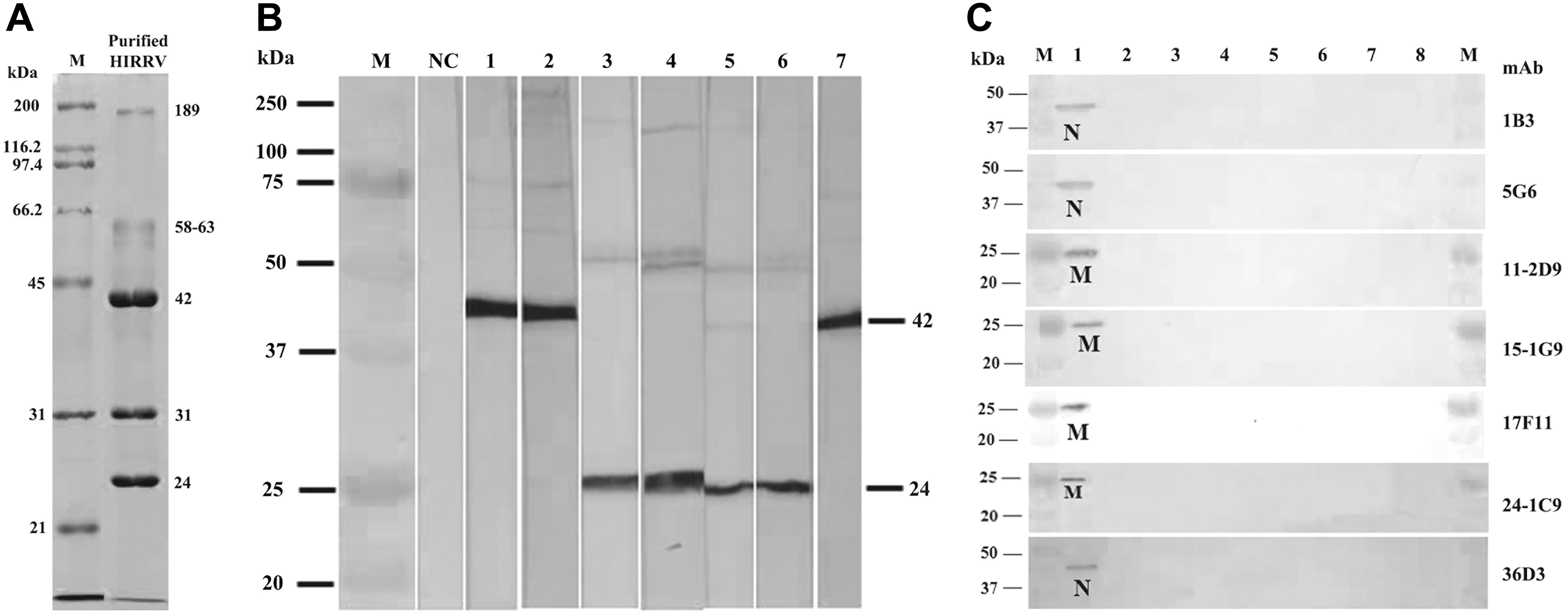
Virus purification and mAb characterization results.
The hybridoma secreting anti-HIRRV mAb were obtained by PEG-assisted fusion of splenic cells of the purified HIRRV immunized mouse and myeloma cells. Resultant antibodies generated from selected hybridoma were screened by ELISA with purified HIRRV, wherein 15 samples gave a positive result. Among positive results, seven hybridoma having a high reaction profile in ELISA were selected and cloned by limited dilution. Finally, we produced seven anti-HIRRV mAbs (1B3, 5G6, 11-2D9, 15-1G9, 17F11, 24-1C6, and 36D3). Isotype determination showed that the heavy chain of all the mAbs except for 5G6 have belonged to the class IgG1, whereas 5G6 has a heavy chain of IgG2a class (data not shown). The L-chain was identified as κ in all seven mAbs.
Characterization and evaluation of anti-HIRRV mAbs
The recognition site and the specificity of seven mAbs were determined using western blot against purified HIRRV, HIRRV-infected EPC cells, and other seven types of fish virus-infected cells, including the normal EPC cells. Characteristically, mAbs 1B3, 5G6, and 36D3 recognized a 42 kDa protein of purified HIRRV, whereas the other mAbs 11-2D9, 15-1G9, 17F11, and 24-1C6 were recognized 24 kDa protein (Fig. 1B). Besides, the produced seven mAbs were able to respond specifically to HIRRV-infected cells and resulting in bands of size 42 and 24 kDa as shown in the Figure 1B (Fig. 1C). No cross-reactivity was observed against EPC cells and six other types of fish virus-infected cells viz. VHSV, IHNV, SVCV, IPNV, MABV, and NNV (Fig. 1C).
Similarly, the least sized structural protein of HIRRV was identified as M protein in all the structural protein elucidations and the identified size of M protein lies in the range of 19.9 to 22 kDa.4,6,19,27 Besides, the most intensively stained protein band in SDS-PAGE was N and its estimated size is in between 42.5 and 46.4 kDa.4,6 On comparison with our SDS-PAGE results, last resolved 24 kDa band is apparently similar with the estimated M protein size and the most intensively stained 42 kDa band was almost identical size of N protein. Therefore, it must be considered that the identified 24 and 42 kDa proteins might be the M and N proteins of purified HIRRV, respectively.
The specificity of the seven mAbs was also determined using ELISA against two HIRRV isolates and six different other fish viruses, including supernatant of EPC cells and DMEM10 (Fig. 2). On comparison of ELISA values, all mAbs against HIRRV isolates showed significantly high OD values than that of the reaction with other viruses. In detail, ELISA values for the isolate HIRRV CA-9703 ranged from 0.53 to 0.67 (0.61 ± 0.02) and that for the second isolate HIRRV Kor-TY15 ranged from 0.24 to 0.52 (0.42 ± 0.03). However, the overall reaction intensity of all antibodies against HIRRV Kor-TY15 isolate was slightly lower compared to that of HIRRV CA-9703 isolate.
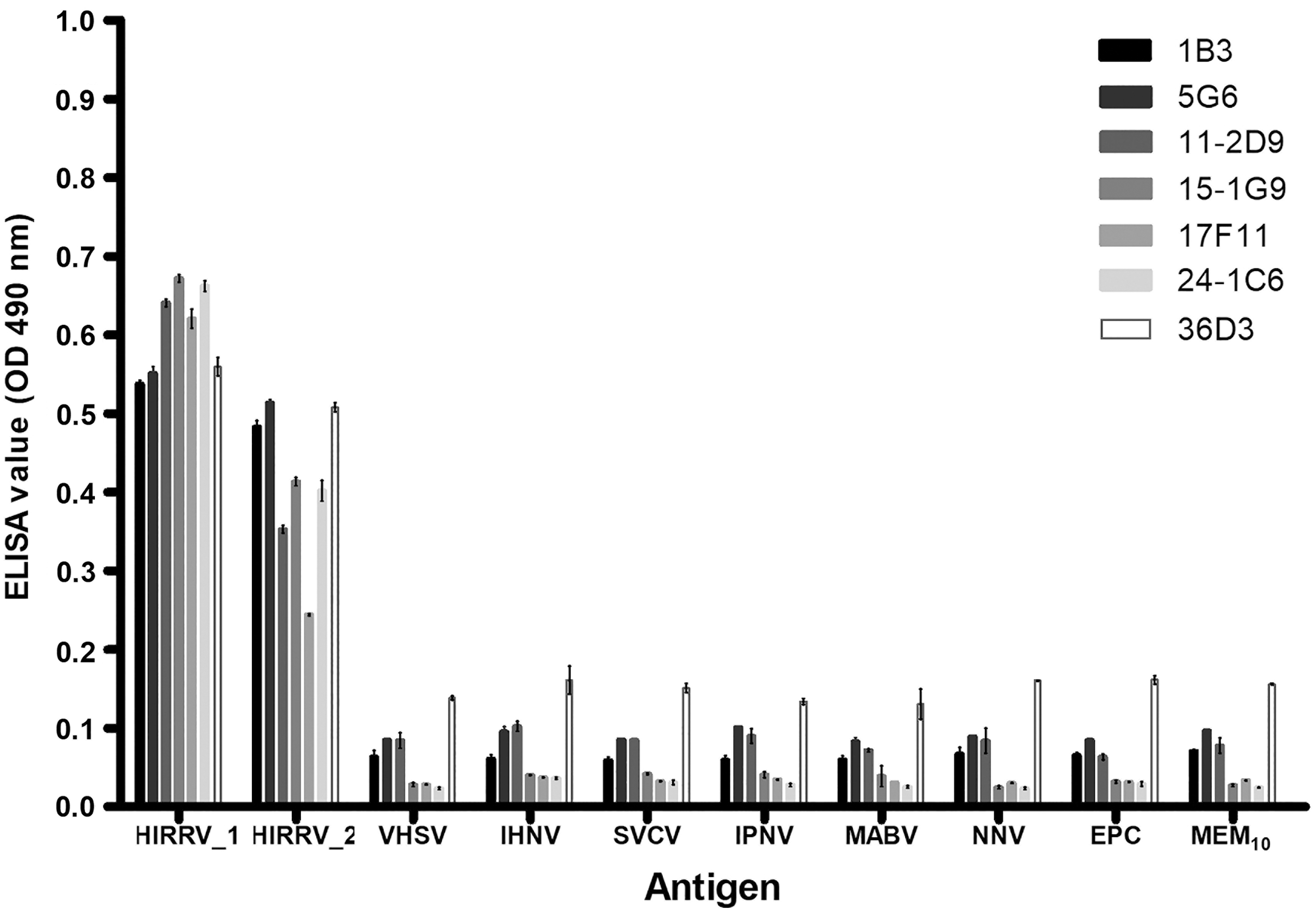
Specificity of seven anti-HIRRV mAbs (1B3, 5G6, 11-2D9, 15-1G9, 17F11, 24-1C6, 36D3) using ELISA against two HIRRV isolates and six different other fish viruses, including supernatant of EPC cells and DMEM. (Amount of antigens:- HIRRV 1: CA9703, 108.3 TCID50/mL; HIRRV 2: Kor-TY15, 107.55 TCID50/mL; VHSV, 109.3 TCID50/mL; IHNV, 107.3 TCID50/mL; SVCV, 107.55 TCID50/mL; IPNV, 109.55 TCID50/mL; MABV, 109.3 TCID50/mL; NNV, 108.05 TCID50/mL; Where, TCID50 indicates the 50% tissue cultures infectious dose). Error bar indicates the SEM. DMEM, Dulbecco's minimum essential medium; ELISA, enzyme-linked immunosorbent assay; SEM, standard error mean.
It can be either due to the difference in the dose of antigen present in the cell culture supernatant (CA-9703: 108.3 TCID50/mL; Kor-TY15: 107.55 TCID50/mL) used or due to the antigenic epitope level variations yielded by a difference in amino acid composition. Moreover, the amino acid sequence homology of P-gene between the isolates CA-9703 (GeneBank accession no. AF104985) and Kor-TY15 (GeneBank accession no. KR011954) was only 94.3% 7 , which might support the latter except 36D3 and showed OD values of <0.11 (0.055 ± 0.01) against other antigens, including six other different fish viruses, EPC cell supernatant, and DMEM10. Similarly, the other six different viruses showed a similar response pattern to those of DMEM10 and EPC cell supernatant (ELISA negative controls) and this could be suggested as a background reaction in ELISA.
To substantiate the results whether the developed mAbs could specifically recognize HIRRV, the indirect fluorescent antibody technique (IFAT) of HIRRV-infected cells using the hybridoma supernatant of mAbs was accomplished. All the seven mAbs produced a similar level of strong green fluorescence signals, which were observed in the cytoplasm of the HIRRV-infected cells (Fig. 3). No positive fluorescence was observed in the uninfected cells as well as IHNV- and VHSV-infected cells, whereas signals were produced as expected in the positive controls against IHNV- and VHSV-infected cells reacted with anti-IHNV and anti-VHSV mAbs (data not shown).

Reactivity of anti-HIRRV mAb to HIRRV-infected cells as assessed using indirect fluorescent antibody technique (IFAT).
As pointed out in the reports,1,6 anti-HIRRV polyclonal antibody (pAb) has cross-reacted with G, N, and M proteins of IHNV and G protein of VHSV, although on a small scale. Therefore, it was suggested that the HIRRV and aforementioned homologous viruses shares a considerable number of epitopes. However, our western blot, ELISA, and IFAT results showed that such cross-reactions have been observed not only against IHNV and VHSV, but also against other fish viruses. Possibly because the developed mAbs might be interacted only with a specific epitope, which is unique in HIRRV and not with a common antigenic determinant contained in other homologous viruses, even though different genotypes of virus are not tested.
Conclusion
Our results strongly indicate that these mAbs specifically recognize HIRRV with no cross-reactivity. The mAbs can be useful and could offer applications in the development of rapid immunodiagnostics for HIRRV considering their ability to specifically detect the HIRRV structural proteins (N and M). Since all HIRRV isolates so far have shown genetic variations from each other,4,7,9–11 the specificity of these developed mAbs must be tested against all available isolates. Therefore, we intend to do more studies on the characterization of mAbs with available isolates of HIRRV.
Footnotes
Acknowledgment
We thank the Ministry of Ocean and Fisheries, Korea and the Indian Council of Agricultural Research (ICAR), New Delhi, India for the support and infrastructure in research.
Authors' Contributions
All persons who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the article. Furthermore, each author certifies that this material or similar material has not been under consideration or published in any other publication before its appearance in the Monoclonal Antibodies in Immunodiagnosis and Immunotherapy. S.K.U. and W.-S.K. designed this study and wrote the article. S.K.U., M.-S.J., and K.-H.K. performed the experiments and analyzed the data. M.-J.O. and W.-S.K. supervised the work and mobilized the funding and resources. All authors read and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by Korea Institute of Marine Science & Technology Promotion (KIMST) funded by the Ministry of Oceans and Fisheries (20150259). The authors also acknowledge the Indian Council of Agricultural Research (ICAR), New Delhi, India for providing overseas doctoral fellowship [NS-ICAR-IF; F.NO. 18(01)/2017-EQR/Edn] to S.K.U.