
Abstract
This Long-Covid disease, mild or severe, is multiorgan or system-wide, spanning from fatigue to clotting abnormalities and autoantibody. The spectrum of different symptoms in Long-Covid diseases makes it difficult to point to a common immunopathogenic etiology. Different immune pathways are presented and critically evaluated. A hypothesis is advanced that indicates autoimmune reactions as cause for Long-Covid disease. The immune network pathway describes a redirection of the nominal anti-SARS-CoV response towards an autoimmune target. Several therapeutic interventions are suggested to suppress the autoimmune pathway.
Introduction
A significant number of SARS-CoV infected patients, around 10%, have persistent symptoms that impact their life. This Long-Covid disease, mild or severe, is multiorgan or system-wide, spanning from fatigue to clotting abnormalities and autoantibody caused diseases.1,2 A recent study on fatigue syndrome reported a persistence of up to 20 months. 3 Interestingly, persistent symptoms are not associated with specific B- or T-cell immunity. 4 Reports on vaccine-induced side effects that resemble Long-Covid symptoms 5 providing evidence for brain-binding autoantibodies associated with neurological symptoms. 1 The role of autoantibodies in long-covid syndrome6,7 challenge the proposed molecular mimicry to explain autoimmunity. 8
Here, I focus on autoimmune reactions leading to Long-Covid symptoms and propose a pathway of idiotypic interactions to understand the cause of Long-Covid syndrome.
The Hypothesis
The view of the immune system as a network was pioneered by Jerne in 19747. Support for a biologically functional system came from several observations:
Antibodies against antibodies, so-called anti-idiotypic antibodies can suppress antigen-specific immunity.9,10 Anti-idiotypic antibodies induce specific immune immunity.6,16–19 The cause of Long-Covid disease has been attributed to a dysfunctional immune system
20
whereby autoantibodies attack critical organ systems.7,18,21–27
Since polyspecific antibodies are extremely rare, 28 they are not considered in as cause for autoimmune reactions in Long-Covid patients. Furthermore, molecular mimicry has been proposed to explain the induction of autoantibodies casing Long-Covid disease, but structural evidence is missing. 8 Evidence is accumulating that autoantibodies are detected in different Long-Covid patients involving various organs.1,29–32 The hypothesis poses that autoantibody cause disease sequala in SARS-CoV infection defeating Paul Ehrlich’s Horror Autotoxicus. The hypothesis is based on a “secondary” immune response observed in sera of repeatedly immunized mice 11 and by B-cells in culture producing antiphosphoryl choline (PC) antibodies and anti-anti-PC. 12 These and subsequent observations of autoantibodies against the first induced response33–35 support the concept of a symmetrical autoantibody response. 36 The symmetrical response is directed against an autoantigen in the V-region of the Ig structure, commonly referred as idiotope. 37 Since the expression of the autoantigenic idiotope is structurally distant to the antigen binding site, it is possible that idiotopes are present on antibodies with different specificities. Idiotopes shared among antibodies with different specificities was discovered in 1971 providing evidence of autoanti-idiotypic antibodies during an immune response, described as Ab1-Ab2. 38
Linked immune responses in Long-Covid symptoms Guillain-Barre Syndrome 39 are cartooned in Figure 1A. The pathway of the symmetrical Ab1-Ab2 responses shows B-cells with receptors (BCR) recognizing the Spike protein produce Ab1. Ab1 expresses an idiotope that is recognized by anti-Ab2 BCR. A subpopulation Ab2 B-cells with the idiotope BCR produce autoantibody against the myelin sheath. This autoantibody damages the protective covering preventing signal transmission by the nerve.
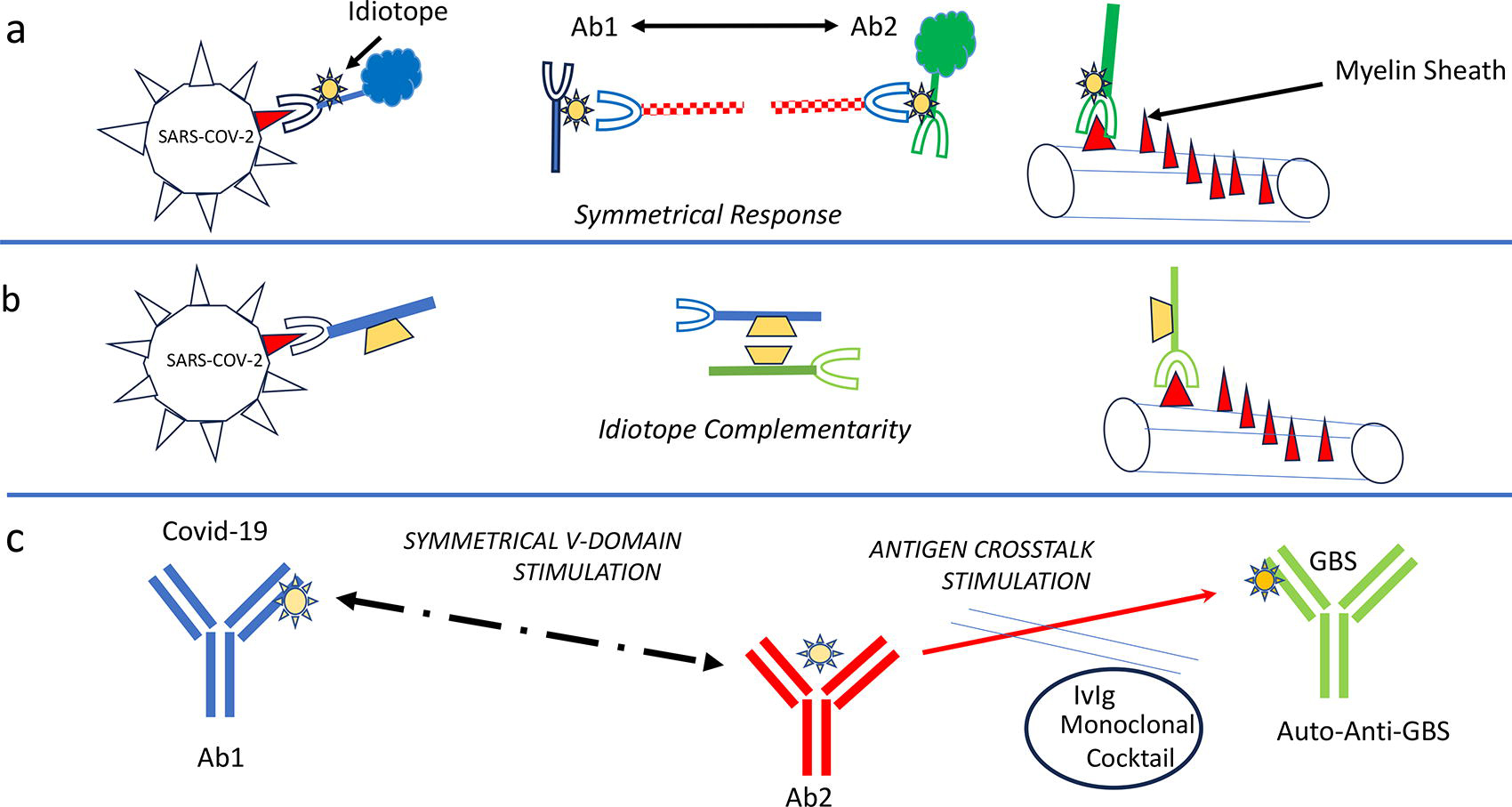
Pathways of Immune Jumping.
Autoimmune antibodies have been demonstrated in several Long-Covid diseases.9–13 The switch from the anti-SARS-CoV response towards an autoantigen contradicts the paradigm the specific adaptive immunity.
Figure 1B shows a hypothesis invoking a different pathway of the immune network. Certain antibodies have variable regions that induce self-binding complementary of V-domain structures have been described in self-binding antibodies. The interaction of V-domain structure is independent of the canonical antigen binding site and can been transferred as so-called homophilic domain. 41 Similar Fab-Fab binding antibodies have been reported. 42 It is conceivable that homophilic regions are expressed by antibodies with different specificities. Antispike antibodies interact with homophilic regions expressed by BCR producing autoantibodies causing Guillain-Barre Syndrome (GBS).
Discussion
Jerne’s network theory 43 impacted the understanding of the immune system recently summarized in the promise of the idiotypic network. 44 A follow-up analysis of the clinical experience using antiidiotypic antibody as medicine yielded a disappointing result. 45 The SARS-CoV epidemy with short-term and long-term diseases kindled renewed interest in the idiotypic network.46–51
About 10% of SARS-CoV infected patients have persistent symptoms that impact their life. This Long-Covid disease, mild or severe, is multiorgan or system-wide, spanning from fatigue to clotting abnormalities and autoantibody caused diseases. 2 A recent study on fatigue syndrome indicate a persistence of up to 20 months. 3 Interestingly, persistent symptoms are not associated with specific B- or T-cell immunity. 4 Recent reports on vaccine-induced side effects have reported that resembles Long-Covid symptoms. 5 These cases however seem to be quite rare. 52 The spectrum of different symptoms in Long-Covid diseases made it challenging to point to a common immunopathogenic etiology. The basis of the proposed pathway are data showing that autoimmune reactions are associated with Long-Covid symptoms. In the proposed hypothesis autoimmune responses are caused by idiotypic interactions. As idiotopes are shared by antibodies against different targets the initial antiviral response can switch to antiself-targets, called “immune Jumping”. This hypothesis provides new insight in the pathology of Long-Covid offering new strategies for therapy. The hypothesis based on the concept of symmetrical immune response: Ab1-Ab2 11 needs to be strengthened experimentally.
Future Directions
In Figure 1C strategies to suppress the generation of auto-autoantibodies are described.
The symmetrical Ab2 response, causing crosstalk autoantibody stimulation is the target to inhibit the autoimmune reaction. Autoimmune reactions can be suppressed using monoclonal antibodies, for example anti-CD20 monoclonals, to eliminate B-cells producing antibodies. 53 A network targeting approach is the use of intravenous immunoglobulin to inhibit autoimmunity, such as GBS, and outlined 40 previously. Finally, a more autoantigen specific intervention would be a cocktail of monoclonal antibodies against the Ab2 response. This cocktail could contain monoclonals against Ab2 from different donors with Long-Covid autoimmune disease. This possibility of producing such monoclonal anti-Ab2 has been demonstrated as well as the immune-modulatory potential in HIV-1 and hepatitis infection.54,55 It is tempting and dogma challenging to extend the Long-Covid specific hypothesis to autoimmune reaction in general as network disease hypothesis.
Conclusion
The finding of changing the antigen target during an immune response violates the dogma of the unique antigen specificity in the adaptive immunity. Antigen immune shifts have been reported as early as 1971. 38 It is hypothesized that autoimmune reactions observed in Long-Covid disease are caused by derailed immune network pathways. This pathway describes the sequala of autoantibodies in Long-Covid immune jumping. The pathway of redirecting the immune target, depicted in Figure 1C, offers therapeutic interventions.
Footnotes
Author Disclosure Statement
The author declares no conflict of interest.
Funding Information
The author has not received funding for the writing of the article.