
Abstract
A loop-mediated isothermal amplification (LAMP) method was developed for the rapid and sensitive detection of the emerging resistance gene New Delhi Metallo-β-lactamase-1 (NDM-1), with its specificity and sensitivity having been evaluated. Six primers, including a pair of outer primers, a pair of inner primers, and a pair of loop primers, were specially designed for recognizing eight distinct sequences on the target NDM-1 gene. The amplification reaction was performed within only 40 min under isothermal conditions at 65°C in a regular water bath. The LAMP assay showed good specificity and higher sensitivity than the conventional polymerase chain reaction (PCR), with a detection limit of 1 pg genomic DNA per tube of one NDM-1-positive reference strain. The detection result for the 345 clinical samples showed 100% consistence with the result by the PCR method, and three contaminated samples could be detected correctly by LAMP assays, while they could not be detected by PCR. The LAMP method reported here demonstrated a potential and valuable means for detection of the NDM-1 gene: easy, rapid, visual, specific, accurate, and sensitive, especially useful for on-the-spot investigation.
Introduction
The conventional methods such as polymerase chain reaction (PCR),3,14 real-time PCR,4,7,12 and DNA microarray, 11 have been successfully used to detect the NDM-1 gene. A novel nucleic acid amplification method, designated loop-mediated isothermal amplification (LAMP), had been well established and documented.10,13 This method relies on an auto-cycling strand displacement DNA synthesis performed by the Bst DNA polymerase, with four to six primers recognizing 6∼8 distinct regions of the target gene and generating the loop-mediated amplification under isothermal conditions ranging from 60°C to 65°C for about 60 min, resulting in large amounts of amplification products with many types of structures. LAMP is more specific, rapid, and simple to perform than PCR. Furthermore, gel electrophoresis is not required, because the LAMP method synthesizes large amounts of DNA where products can easily be detected by turbidity or fluorescence. Therefore, the LAMP assay has emerged as a powerful tool that facilitates genetic testing for the rapid diagnosis of infectious diseases, including bacteria and virus identification.2,5
This study aimed at developing and evaluating a simple and rapid detection method based on LAMP assays for the NDM-1 gene and applying these assays to detect the NDM-1-positive strains from various samples. As far as we know, this is the first report of a rapid, specific, and sensitive method for the detection of the NDM-1 gene by LAMP assays.
Materials and Methods
Bacterial strains and genomic DNA extraction
One NDM-1-positive reference strain (Acinetobacter lwoffi) was isolated from a chicken farm in the Shandong Province of China and was used to optimize the LAMP reaction system, including the temperature, the time, and the sensitivity. Four standard bacteria, including E. coli (ATCC 25922), Salmonella enterica (ATCC 50035), Staphylococcus aureus (ATCC 292130), and Pseudomonas aeruginosa (ATCC 27853); the NDM-1-positive reference strain; and 50 known clinical strains were used to evaluate the specificity of the LAMP assays. The 345 other clinical strains were included to apply the established LAMP assays for detection. The 395 clinical strains were isolated by the swabs of anuses from chicken and pigs and preserved in our laboratory, including E.coli, Klebsiella pneuminiae, and Pseudomonas aeruginosa isolates, as well as Acinetobacter baumannii isolates. All of them showed resistance to imipenem in the phenotype. Meanwhile, the imipenem resistance-related gene VIM and IMP were tested by PCR methods. Totally, 236 clinical strains were VIM positive, 65 clinical strains were IMP positive, and the other 94 strains were not detected, suggesting the possible mechanism of resistance mediated by efflux or impermeability or unknown factors.
For genomic DNA extraction, a single clone of the bacteria was picked from a Luria–Bertani agar plate and incubated at 37°C for 12∼26 hr to obtain 108 CFU/ml in Luria–Bertani broth. One milliliter aliquots of the enriched culture were centrifuged at 12,000 rpm for 2 min, and the precipitation was used for the genomic DNA extraction according to the manual of Bacteria Genome Extraction Kit (Tiangen Biotech Co., Ltd.). The extracted genomic DNA was diluted in distilled water (DW) to a final volume of 50 μl to be used as template for the LAMP and PCR amplification. The concentration of the genomic DNA of each isolate is measured by Nanodrop 2000 (Bio-Rad).
Primers design
The LAMP and PCR primers for detection of the NDM-1 gene are given in Table 1. A set of inner primers (including forward inner primers [FIP] and backward inner primers [BIP]), outer primers (including F3 and B3), and loop primers (including LF and LB, to accelerate reaction) were specially designed for the LAMP reaction to target eight distinct regions, as shown in Fig. 1. The primers were designed using the PrimerExplorer V3 software (http://primerexplorer.jp/e/) and commercially synthesized by Invitrogen Biotech). The PCR primers were designed using the Primer 5.0 software, allowing the amplification of a sequence of 390 bp. The NDM-1 gene sequences were taken from the DNA sequence of E. coli megaplasmid (GenBank accession No. AB571289.1), K. pneumoniae plasmid pKpANDM-1 sequence (GenBank accession No. FN396876.1), K. pneumoniae strain ADB65 plasmid (GenBank accession No. HM853678.1), and K. pneumoniae strain CMC Micro VB1 (GenBank accession No. HQ171206.1).
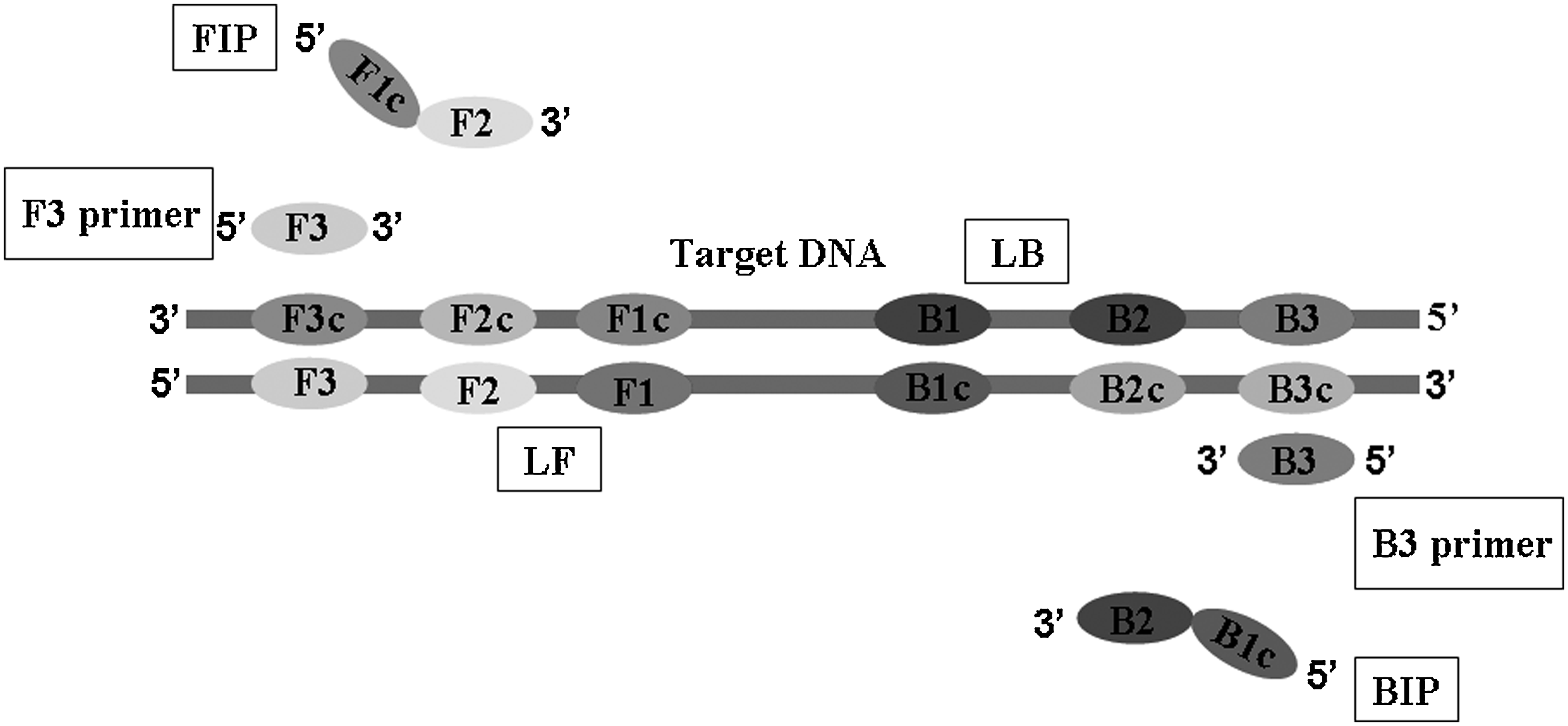
Schematic representation of LAMP primers. The Inter primers were FIP and BIP. FIP consisted of the complementary sequence of F1 and F2. Inter Primer BIP consisted of B1c and the complementary sequence of B2c. The outer primers were F3 and B3, which were located outside of the F2 and B2 regions, respectively. The loop primers LF and LB were located between F2 and F1 or B1 and B2, which were designed to anneal at the loop structure of the amplicons and accelerate and enhance the sensitivity. LAMP, loop-mediated isothermal amplification; FIP, forward inner primers; BIP, backward inner primers; LF, forward loop primers; LB, backward loop primers.
F3, forward outer primers; B3, backward outer primers; FIP, forward inner primers; BIP, backward inner primers; LF, forward loop primers; LB, backward loop primers; NDM-F, forward PCR primers; NDM-R, reverse PCR primers; LAMP, loop-mediated isothermal amplification; PCR, polymerase chain reaction.
Analysis of LAMP and amplification products
The LAMP assays were carried out in a total of 25 μl reaction mixture containing 40 pmol (each) of the primers FIP and BIP, 5 pmol (each) of the primers F3 and B3, 20 pmol (each) of the loop primers LF and LB, 2×Reaction Mix [40 mM Tris-HCl(pH8.8), 20 mM KCl, 16 mM MgSO4, 20 mM (NH4)2SO4, 0.2 wt% Tween20, 1.6 M betain (Sigma) and 2.8 mM dNTP] 12.5 μl, 1.0 μl (8U) of Bst DNA polymerase (New England Biolabs), 1.0 μl Fluorescent Detection Regent/FD (Eiken Chemical Co., Ltd), and 2.0 μl target genomic DNA; DW was added to the others. First, the reaction tube was incubated at 60°C, 63°C, and 65°C by adding inner primers FIP/BIP and outer primers F3/B3 for 60 min in the Loopamp Real-time Turbidimeter LA-320C (Eiken Chemical Co., Ltd; Terasaki Electric Co., Ltd.) for screening out the optimal temperature and on the basis of the temperature, we performed the LAMP assays together with the loop primers LF/LB to ensure the shortest time for amplification.
LAMP assays have the characteristic of high amplification efficiency, and there are two methods for determination of the positive and negative judgment. On the one hand, without the FD adding tube, the existence of the target DNA can be confirmed by checking, whether there is white turbidity caused by the existence of the amplification by-product magnesium pyrophosphate. On the other hand, with the FD adding tube, LAMP amplifications in the reaction mixture were generally observed by naked eyes or UV light on the color change. The color of the solution turned obvious green in the presence of LAMP amplification, while it remained orange without any amplification. For further specificity confirmation, LAMP products were digested with 5 U of restriction enzyme DdeI (New England Biolabs). The original amplicons and digested products were subjected to electrophoresis at 2% agarose gel.
Comparison of sensitivity and specificity between PCR and LAMP assays
In order to compare the sensitivity of the LAMP assays, the genomic DNA of the NDM-1-positive reference strain was serially diluted 10-fold with sterile DW, ranging from 10−14 to 10−7 g DNA, respectively. A negative control was performed using sterile DW. PCR was performed with the NDM-F/NDM-R primers (Table 1). The amplification was carried out in a 25 μl total reaction volume with 1 μl genomic DNA as template, 5 μl of 10×PCR Buffer (100 mM Tris-HCl, 500 mM KCl, 15 mM MgCl2, pH 8.3), 2.0 μl (10 μM) of a couple of appropriate primers (NDM-F/NDM-R), 2.5 μl of dNTPs mixture (2.5 mM of each dNTPs), and 0.25 μl (5U/μl) of Taq DNA polymerase (TaKaRa Bio). The thermal profile for PCR was 95°C for 10 min, followed by 35 cycles of 95°C for 1 min, 60°C for 45 sec, and 72°C for 45 sec and a final extension cycle at 72°C for 10 min. For comparison, LAMP assays were performed using the same diluted genomic DNA as templates by the optimal condition. The LAMP and PCR amplification products were analyzed by electrophoresis at 2% agarose gel.
To evaluate the specificity, four standard bacteria, 50 known clinical strains, and the NDM-1-positive strain were used for testing by PCR and LAMP assays under optimal conditions, respectively. All the LAMP products were digested by DdeI for confirmation of the specificity. These experiments were replicated to ensure reproducibility.
Application of LAMP assays on clinical isolates
The genomic DNA of the 345 clinical isolates from chicken and pigs and the NDM-1-positive reference strain was prepared through the same kit just discussed, and was then subjected to detection by LAMP and PCR assays as mentioned earlier. The LAMP products were determined by the turbidity or the color change, while the PCR products were evaluated by electrophoresis as just mentioned. These experiments were replicated to ensure reproducibility.
Results and Discussion
Optimization of the conditions of LAMP assays and LAMP products analysis
In order to determine the optimum conditions of the LAMP assays, 100 ng genomic DNA from the NDM-1-positive reference strain was used as template. LAMP assays were performed under isothermal conditions at 60°C, 63°C, and 65°C, respectively, when only using inner primers FIB/BIP and outer primers F3/B3; the amplification products could not be observed until 40 min, and no significant difference was observed at three different temperatures; however, the LAMP products at 65°C showed the highest level of amplified amount of the target gene. Furthermore, after adding the loop primers LF/LB at 65°C, the amplification was initially detected at about 20 min, and the reaction time was accelerated at least 15 min, indicating that the loop primers played an important role on the speed of the amplification. The reaction time of the LAMP assays was determined to be 40 min according to the appearing time of the positive results with the minimum template amount. In our study, we designed the loop primers as well as the different inner and outer primers, so the reaction time was greatly shortened from 1 hr to 20 min compared with the established LAMP method. 15 Moreover, the positive template was closer to clinical reality, because the NDM-1-positive reference strain (A. lwoffi) was used, while the NDM-1 gene by artificial synthesis was used in the method just mentioned.
The LAMP products were analyzed by observation of the reaction mixture by either the naked eye or 2% agarose gel electrophoresis. The positive results were shown as in Fig. 2A tube 1 and 3, and the negative results were shown as in Fig. 2A tube 2 and 4. The typical LAMP laddering pattern was observed, indicating various replicating intermediates of the stem loop amplification process, while the negative control has no amplification (Fig. 2B). The digestion of LAMP products with the DdeI restriction endonuclease resulted in two DNA fragments (Fig. 2C), approximated at 75 and 117 bp, similar to the predicted sizes. Therefore, the NDM-1 gene segment was specifically amplified.

Analysis of LAMP products and the digestion products.
Specificity and sensitivity between LAMP and PCR assays
The LAMP assays correctly detected the NDM-1-positive reference strain, while 50 other known clinical isolates (including 25 VIM-positive strains, 8 IMP-positive strains) and four standard bacteria showed negative results with no amplification. The LAMP products were digested by DdeI, and the result showed that the positive LAMP product exhibited the same two digestion bands similar to the predicted sizes. The results were consistent with those using PCR through NDM-F/R primers (data not shown), indicating the good specificity of the established LAMP assays. In fact, until now, we have found only this one NDM-1-positive strain in veterinary clinics of China.
The detection limit of LAMP assays was 1 pg genomic DNA/tube (Fig. 3A, B), and PCR sensitivity was determined to be 10 pg DNA/tube (Fig. 3C), indicating that the LAMP assay was at least 10-fold more sensitive than the PCR method. The result was in accordance with the previous research. 16
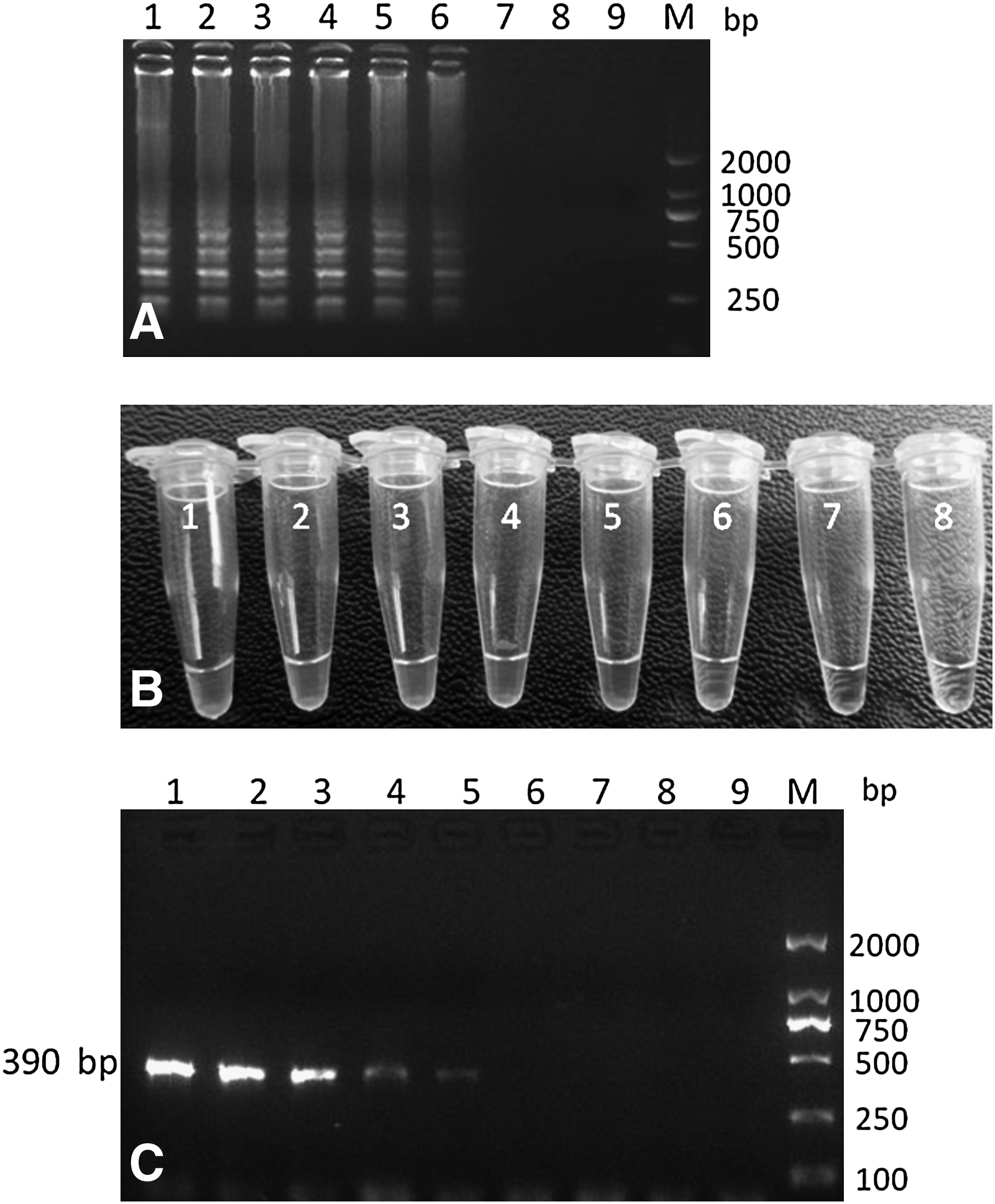
Sensitivity between LAMP and PCR assays.
Application of LAMP assays to clinical isolates
The established LAMP assays were applied to detect 345 clinical isolates (including 211 VIM-positive strains and 57 IMP-positive strains) from chicken and pigs in a water bath at 65°C for 40 min. For comparison, the 345 samples were analyzed by PCR as mentioned earlier. The result showed that only the NDM-1-positive reference strain was detected to be positive by LAMP assays, and others were negative; meanwhile, no false positive was observed, which was consistent with the result by PCR (data not shown). Due to the lack of positive samples for testing the application of the LAMP assays established in our study, we contaminated three clinical genome samples with 10, 1, and 0.1 pg of the positive-NDM-1 A. lwoffi genome DNA, respectively. The results showed that LAMP assays could detect all these three contaminated samples, while PCR could not, indicating that LAMP is indeed better than PCR for rapid detection of the NDM-1 gene in clinics.
Conclusion
Horizontal transmission of bacteria resistance is a serious threat, so the rapid supervision of the NDM-1 gene is most important for the health of animals and human beings. 6 The LAMP assays for the detection of the NDM-1 gene demonstrated higher sensitivity and good specificity. The assays consist of the isolation of the clinical isolates and the extraction of genomic DNA, which is finally analyzed by the LAMP assays for the detection. In the present study, the detection limit of this LAMP assay was 1 pg DNA/tube, while that of the PCR method was 10 pg/tube. The specificity was validated by four standard bacteria and 50 known clinical strains as well as the NDM-1-positive reference strain. The diagnostic accuracy for the 345 clinical isolates was 100% when compared with the traditional PCR method. Moreover, the LAMP assays established in this study could detect three contaminated clinical samples with trace NDM-1 genome DNA to be positive, while PCR could not. In addition, the LAMP method is carried out under the isothermal condition; therefore no special apparatus is needed, a regular laboratory water bath or heating block is good enough, which decreases the cost of detection. Most important, the reaction time is greatly shortened, from 2∼3 hr to 40 min. Meanwhile, the result is easy to be determined, merely by observation of the white turbidity or the color change by the naked eye. In conclusion, the LAMP assay has the potential to become a standardized method for the detection of the NDM-1 gene in laboratories and in on-the-spot investigation for its rapidity, sensitivity, specificity, simplicity, and low cost.
Footnotes
Acknowledgment
This work is supported by a grant from the Special Fund for Agro-scientific Research in the Public Interest of China (No. 201203040) and the Natural Science Foundation of China (No. 81171621).
Disclosure Statement
No competing financial interests exist.