
Abstract
Tyrosyl-tRNA synthetases (TyrRSs) as essential enzymes for all living organisms are good candidates for therapeutic target in the prevention and therapy of microbial infection. We examined the effect of various polyphenols, alkaloids, and terpenes—secondary metabolites produced by higher plants showing many beneficial properties for the human organism, on bacterial aminoacylation reaction. The most potent inhibitors of Escherichia coli TyrRS are epigallocatechin gallate, acacetin, kaempferide, and chrysin, whereas the enzymes from Staphylococcus aureus and Pseudomonas aeruginosa are inhibited mainly by acacetin and chrysin. Most of them act as competitive inhibitors. Structure–activity relationship showed that the most potent flavonoid inhibitors contain hydroxyl group at position 5 and 7 of A ring and OCH3 group at position 4′ of B ring.
Introduction
T
Inhibition of one of these two enzymatic steps disrupts tRNA charging, which in turn stalls elongation of growing polypeptide chains. 6 AaRSs are promising targets for development of cures against pathogenic species.7,8 It has been shown that many of the naturally occurring antimicrobials specifically inhibit aaRSs activity.9,10 The unique features that cause these enzymes to be considered as a good therapeutic target are: (i) the differences in structure of prokaryotic and eukaryotic aaRSs; (ii) phylogenetic conservation of aaRS, which causes antimicrobials targeting aaRS from one bacteria have the potential to inhibit homologous enzymes from others; and (iii) the presence of twenty distinct aaRSs in most bacterial species representing independent targets.11,12
Flavonoids, alkaloids, and terpenes are the group of natural compounds, commonly found in food products, with many biological and pharmacological activities. Their antibacterial, antiviral, antioxidant, and antimutagenic effects and inhibition of several enzymes have already been demonstrated. 13 It is believed that because flavonoids are widely distributed in edible plants and beverages and have previously been used in traditional medicine, they are likely to have minimal toxicity. 14 However, this family of compounds show a wide range of activities in mammalian cells in vivo, thus thorough analysis of their side effects is necessary for the complete assessment of their potential for therapeutic utility.15–17 Given that the selectivity of flavonoids with respect to eukaryotic enzymes appears to vary depending on the compound, a study to assess the toxicity of these phytochemicals should be carried out for each of them individually.15,18
In this work, we present the results of our study on dependence between structure–activity of flavonoids as inhibitors of aminoacyl-tRNA synthetase.
Materials and Methods
Strains and plasmids
Escherichia coli LMG194 (Invitrogen) genotype: F-ΔlacX74 galE thi rpsL ΔphoA (Pvu II) Δara714 leu::Tn10 with a deletion of araBADC.
Staphylococcus aureus, clinical isolate (Collection of the National Institute of Health, Warsaw, No. 194/10) MRSA, resistant to: erythromycin, clindamycin, doxycycline, gentamicin, ciprofloxacin, trimethoprim/sulfamethoxazole; sensitive to: vancomycin, linezolid, teicoplanin, and rifampicin.
Pseudomonas aeruginosa, clinical isolate (Collection of the National Institute of Health, Warsaw, 1893/09), resistant to: ciprofloxacin, carbapenems (imipenem, meropenem) sensitive to: amikacin, ceftazidime, gentamicin, piperacillin, and netilmicin.
E. coli ATCC 25922, control strain for testing susceptibility to antibiotics ESBL and others. Bacterial media: BBL™ Mueller Hinton II Agar (Becton, Dickinson & Co.). Plasmid pGB2Ω inv/hly (10.05 kb), kindly provided by Prof. Catherine Grillot-Courvalin, (Antibacterial Agents Unit, Department of Microbiology, Institut Pasteur, Paris, France), expressing invasin from Yersinia pseudotuberculosis. Plasmid pAT505 (pUC18Vgfp-mut1, ApR) (3.45 kb) consisting 750-bp XbaI–PstI fragment carrying the gfpmut1 gene encoding green fluorescent protein (GFP) cloned into the XbaI–PstI sites of pUC18 under the control of the Plac promoter.
Expression and purification of tyrosyl-tRNA synthetase
E. coli tyrosyl-tRNA synthetase (TyrRS) coding region was amplified by polymerase chain reaction from genomic DNA isolated from E. coli using the following primers: forward 5′-GGTGGTTGCTCTTCCAACGCAAGCAGTAACTTG-3′; reverse 5′-GGTGGTCTGCAGTCATTTCCAGCAAATCAG-3.′
The PCR product was digested and cloned into SapI/PstI site of pTYB21 (NEB) with fused intein-tag at N-terminus. Correct construct was confirmed by colony PCR using the primers: forward, 5′-CCCGCCGCTGCTTTTGCACGTGAG-3′ reverse, 5′-GGTGGTCTGCAGTCATTTCCAGCAAATCAG-3′, and by sequencing.
The above construct was then used to transform E. coli ER2566 (NEB).
Transformed bacteria were cultured in LB medium supplemented with 100 μg/ml ampicillin. Protein expression was induced at 0.5 OD600 with 0.4 mM IPTG and carried out for 12 hr at 22°C to increase solubility of the protein. After centrifugation, cell pellet (2.5 g) was suspended in 20 ml of solution containing 20 mM Tris-HCl pH 8.5, 0.5 M NaCl, 1 mM EDTA, and protease inhibitors, sonicated and centrifuged. The supernatant containing protein was supplemented with 0.01% of Triton X-100 to final volume of 100 ml. TyrRS containing N-terminal intein was further chromatographed on Affinity Chitin-Binding Tag Agarose (NEB). The purity of TyrRS was estimated by SDS-PAGE. Protein concentration was determined by Bradford method.
In vitro aminoacylation assay
The reaction was carried out in 50 μl of reaction mixtures containing 40 mM Tris-HCl pH 7.5, 2.4 mM MgCl2, 28 mM KCl, 0.8 mM β-mercaptoethanol, 1.6 mM ATP, 0.4 mg/ml E. coli crude tRNA (Sigma), 2.5 μM [ 14 C]Tyr, 2.5 nM TyrRS, and a range of different concentration of inhibitor. After 30 min of incubation, the reaction was quenched with 100 μl of 10% (w/v) trichloroacetic acid (TCA) and kept on ice for 10 min. Aliquots were spotted on 3MM Whatman filter disks presoaked in 10% TCA, washed twice with 5% TCA, and 95% ethanol. The filter was then dried out, and the radioactivity was counted using scintillation counter (MicroBeta; PerkinElmer). Data were analyzed with the FindGraph software.
Determination of antimicrobial activity
For disc diffusion analysis, Nutrient MHII Agar plates were seeded with 24 hr broth culture of different bacteria suspended in physiological solution of NaCl to 0.5 McFarland turbidity standards. In each of these plates, four sterile filter discs of 6 mm diameter soaked with 20 μl of 50 mM solution of analyzed compound were placed. The plates were then incubated at 35°C for 24 hr. The antimicrobial activity was evaluated by measuring the diameter of clear zone surrounding the filter. Standard discs of the antibiotic were applied as a positive antibacterial control.
Determination of minimal inhibitory concentration
The minimal inhibitory concentrations (MIC50s) of the analyzed compounds were determined by MTT assay. 19 One hundred microliters of E. coli, S. aureus, and P. aeruginosa (105cfu/ml) in LB medium containing ampicillin (50 μg/ml) were seeded in 96-well plate and supplemented with inhibitor dissolved in phosphate-buffered saline/dimethyl sulfoxide (PBS/DMSO) (50% v/v) at final concentrations of 500, 250, 100, and 50 μM/well. As positive and negative controls PBS in DMSO (50% v/v) and 50 μg/ml of kanamycin were used, respectively. After incubation for 24 hr at 37°C 50 μl of PBS containing 2 mg of MTT/ml (methylthiazolyldiphenyl-tetrazolium bromide) was added to each well and left at ambient temperature for 4 hr in the dark followed by centrifugation (10 min/4,000 rpm). Supernatant was carefully removed and the pellets were dissolved in 100 μl of solution containing 10% SDS, 5% isopropanol, and 1 M KCl for 12 hr at ambient temperature. Concentration of Formazan was measured at λ −590 and 620 nm as a reference with a microplate reader (Synergy2; BioTek) at 550 nm. MIC50 was determined from six independent measurements for each inhibitor concentration.
Cytotoxicity of antibacterial compounds
Analysis was carried out using human embryonic kidney cells (HEK293) derived from healthy fetus (Sigma). Around 5 × 103 of cells were seeded per well in 96-well tissue plates and cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma) supplemented with 10% fetal bovine serum (Gibco), 1% antibiotics (Sigma), and 1% MEM vitamin solution (Sigma) and incubated for 24 hr at 37°C under 5% CO2 atmosphere. Then tested compounds at the concentrations: 0.05–1 mM were added and incubated for the next 24 hr. After that, the cells were washed with PBS buffer and 100 μl of fresh medium supplemented with 1 mg/ml of MTT was added to each well. After 4 hr at 37°C supernatants were discarded and 100 μl of DMSO was added. The concentration of Formazan was measured at λ −590 and 620 nm as a reference using a spectrophotometer (Synergy2; BioTek). Specific cytotoxicity (%C) was calculated as follows: %C = 100 – [optical density of effectors + targets] – [optical density of effectors]/[optical density of targets] × 100. 20
Bacterial invasion test
E. coli LMG194 were cultured until 0.7 OD600 in LB medium supplemented with 50 μg/ml of spectinomycin and ampicillin at 37°C. After that, bacteria were transferred to the six-well plate containing different concentrations of inhibitors (500, 250, and 100 μM) and incubated 24 hr at 37°C.
One hundred microliters of the kidney epithelial cells extracted from an African green monkey (VERO cells) suspension (105/ml) were seeded into six-well plates in complete culture Eagle's minimum essential medium (EMEM) supplemented with 10% fetal calf serum and incubated overnight.
Bacteria incubated previously with inhibitor were harvested, suspended in EMEM medium, and added to VERO cells. After 2 hr of incubation at 37°C, the cells were washed three times with EMEM and incubated for 0.5 hr at 37°C in complete medium containing 50 μg/ml of gentamicin to remove extracellular bacteria. VERO cell were then washed three times with EMEM medium and examined under fluorescent microscope (Leica). Internalized bacteria were visible, thanks to fluorescence of GFP.21–23
Docking of acacetin and kaempferide in E. coli TyrRS structure
Docking of the compounds to structures of TyrRS from E. coli and S. aureus was performed using 3D conformers deposited in PubChem data base (kaempferide—CID 5281666, acacetin—CID 5280442).
24
Structure of wild-type E. coli AARS was modeled based on the structure of E. coli TyrRS mutant complexed with 5′-O-[N-(
Results
Thirty-four flavonoids and seven other natural compounds (Table 1) differing in their structure and hydroxyl group contents were evaluated for their in vitro activity in inhibition of aminoacylation reaction catalyzed by TyrRS from E. coli, S. aureus, and P. aeruginosa (Table 2).

C, competitive; M, mixed; N, noncompetitive; NA, not analyzed; U, uncompetitive inhibition.
Inhibition of bacterial TyrRS activity
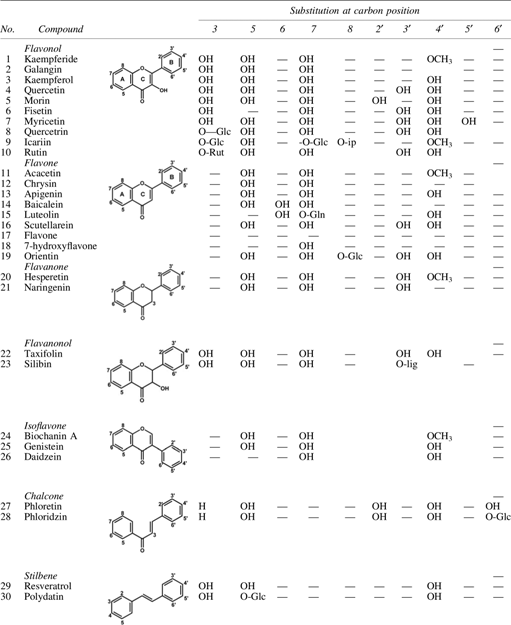
Effects of these compounds on aminoacylation reaction was analyzed using purified recombinant E. coli TyrRS (KM −4.1 μM, Vmax –3.73 μmol/min/mg) and partially purified enzymes from S. aureus and P. aeruginosa (assay details are described in Materials and Methods). Inhibitory activity of analyzed compounds expressed as Ki or IC50 are presented in Table 2 and Fig. 1.
(
Based on aaRS inhibitory activity, polyphenols were divided into three groups: (i) compounds exhibiting less than 10% residual activity at 200 μM concentration: 1, 2, 11, 12, 13, 20, 21, and 24; (ii) compounds exhibiting ∼10–50% residual activity: 3, 4, 5, 14, 15, 16, 25, 27, and 34; and (iii) those exhibiting more than 50% residual activity: 6, 7, 8, and 29 (Fig. 1a). No inhibition at all was detected for 9, 10, 19, 32, and 33 (IC50 ≥ 2 mM). Generally the polyphenolic compounds demonstrated medium activity against E. coli TyrRS with IC50 in micromolar range (Table 2). The most potent inhibitory activity (of all tested in this work) against E. coli TyrRS show compounds 31, 11, 1, 12, 24, and 13 (Table 2). In the case of S. aureus and P. aeruginosa IC50 are ∼10–100 times higher than E. coli. Compounds 27, 12, and 11 inhibit TyrRS of S. aureus at concentrations 100, 146, and 146 μM, respectively, whereas 7 and 12 inhibit TyrRS of P. aeruginosa with IC50 550 and 567 μM, respectively (Table 2). For comparison tested compounds (except of 38 and 35) weakly inhibit eukaryotic TyrRS derived from Sus scrofa domestica (S. scrofa domestica), which homology to human enzyme amounts ca. 80%.
The lowest value of inhibition constant (Ki) of the most active inhibitors are in range of KM for tyrosine (Table 2). KM for Tyr was determined by Lineweaver–Burk plot (E. coli TyrRS KM −4.1 μM) (Fig. 1b). Dixon plot (Fig. 1c) for inhibition of E. coli TyrRS with increasing inhibitor concentrations show straight lines intersected in the II quadrant of the Cartesian plane, which indicate competitive or mixed mode of inhibition of TyrRS by compounds: 1–4, 11–16, 20, 21, 24, 25, 27, and 34 (Table 1). Analysis by Cornish-Bowen plot clearly showed that most of the compounds act as competitive inhibitors, except 16 and 31, which are noncompetitive and 30 and 34, which interact in mixed and uncompetitive modes, respectively (Table 2; Fig. 1d–g). Comparison of Ki′ < Ki means that 30 binds to enzyme–substrate complex.
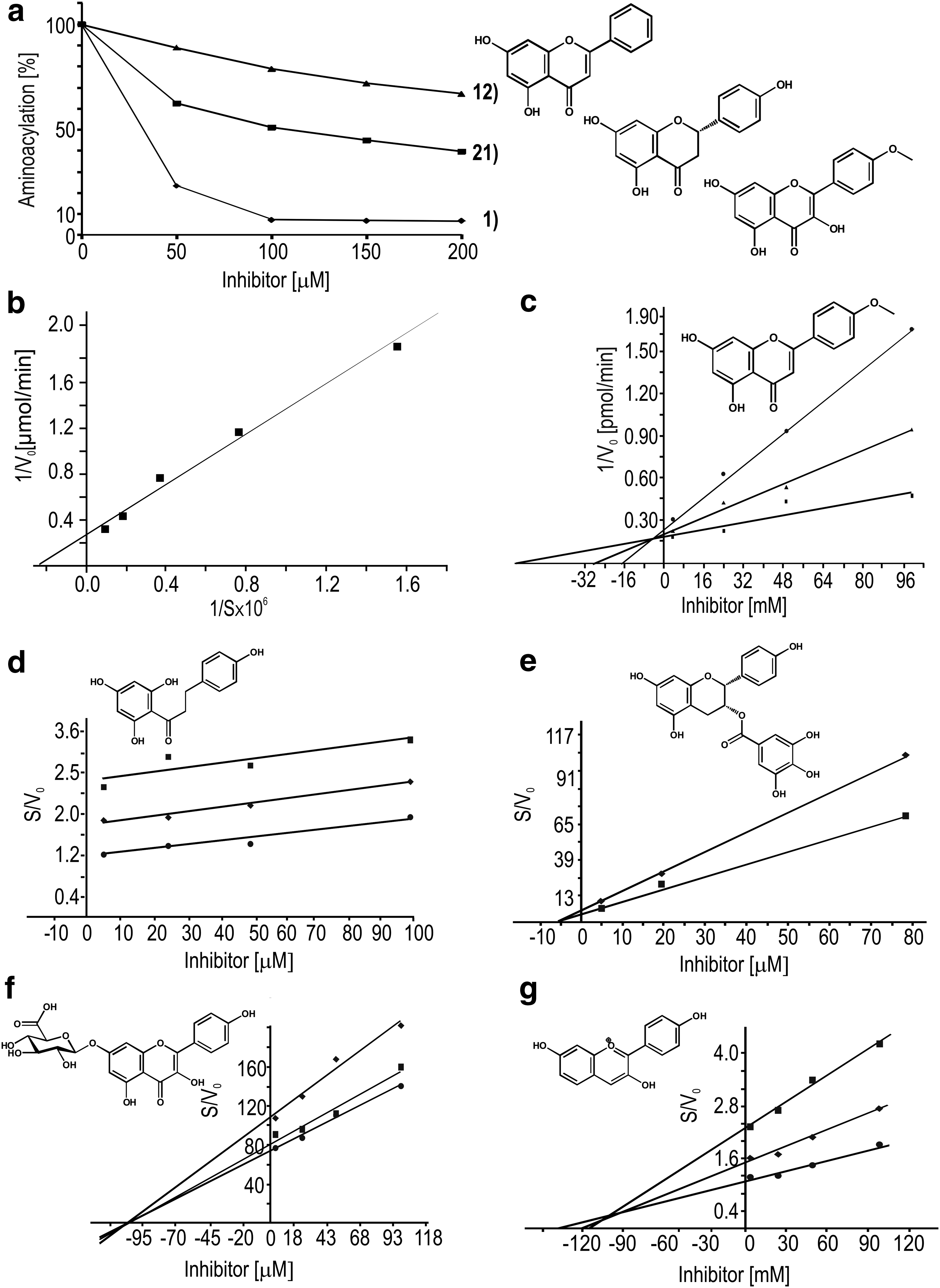
Comparing the effect of 8, 27, and 29 on aminoacylation reaction catalyzed by TyrRS, one can notice that in the presence of ca. 200 μM of the substance reaction is inhibited in 80%, 70%, and 30%, respectively. Substitution of sugar moiety at position 3 or 7 of ring A weakens inhibitory potency of aglycone and changes inhibitory mode as seen in the case of 16. Whereas in the case of 28 (27 substituted at position 5) and 30 (29 substituted at position 7) up to 180 μM enhancing of aminoacylation yield was observed, while further increase of their concentration inhibited TyrRS activity (Fig. 2). Moreover, addition of sugar moiety to 29 leading to the formation of 30, changes the mode of inhibition into mixed (Table 2).
Inhibition of aminoacylation reaction catalyzed with TyrRS in the presence of (
In addition of polyphenols, we analyzed the inhibitory activity of a series of terpenes and alkaloids. The best results were obtained for 36 and 35, which inhibit aminoacylation reaction with Ki 49 and 85 μM, respectively (Table 2). Dixon plot revealed that these compounds act as noncompetitive inhibitors.
Antibacterial activity
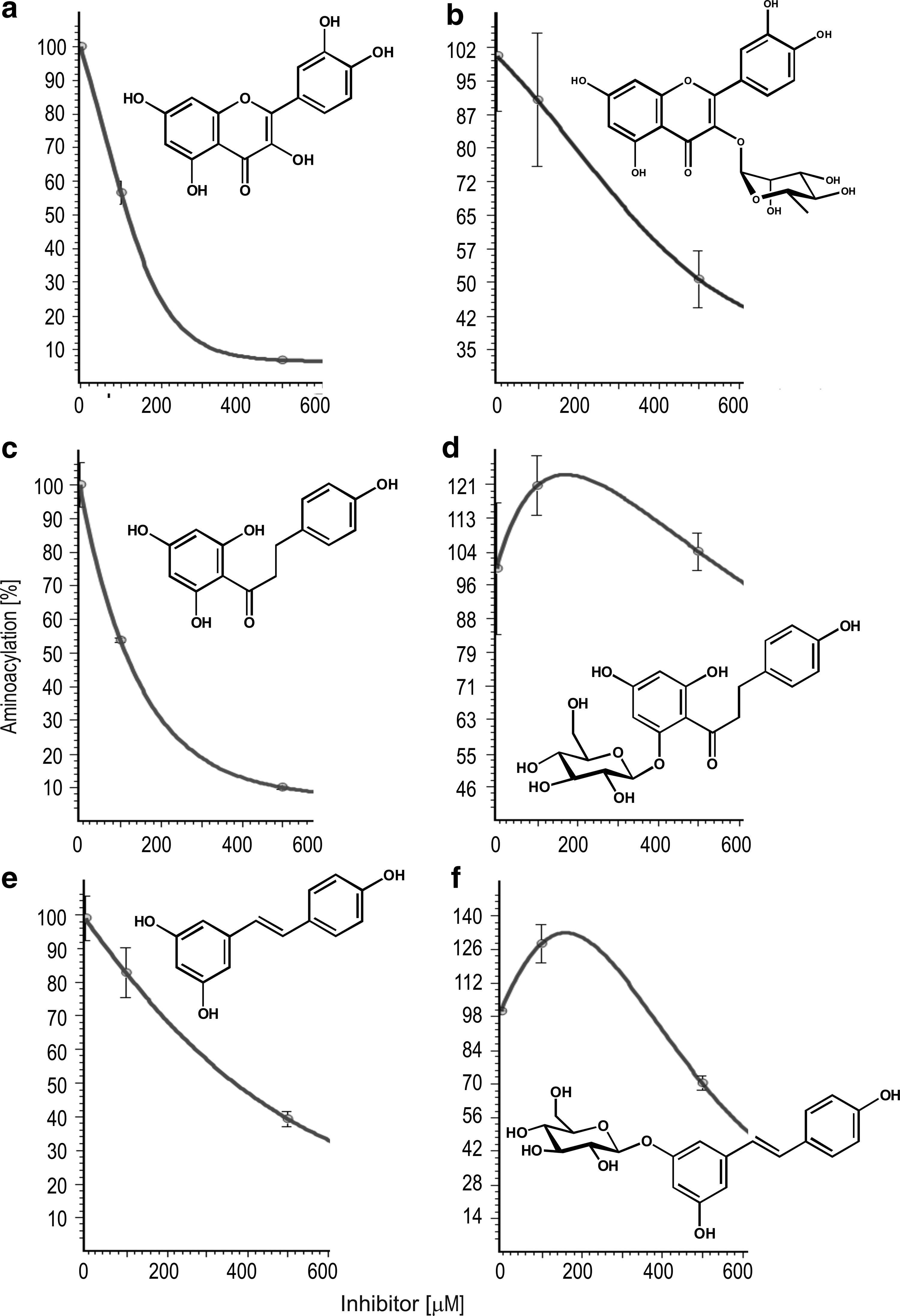
The antimicrobial activity of the most potent aaRS inhibitors against E. coli, S. aureus, and P. aeruginosa was evaluated in in vitro conditions by the broth microdilution method. 27 The results are presented in Table 3. In general, a few evaluated compounds exhibit good activity against S. aureus and slightly weaker against P. aeruginosa (1, 11, 24) and E. coli (4, 34) (MIC50 ranging from 8 to 190 μM). The lowest MIC50 correspond to low IC50 values for inhibition of TyrRS. This positive correlation suggests that their antibacterial activity may be due to inhibition of TyrRS.
Zone inhibition analysis
For S. aureus (MRSA) growth inhibition zone of 19 and 15 mm of diameter was observed only around the disks soaked with 36 and 31, respectively (Fig. 3a, b). 36 also showed some growth inhibition of E. coli strain (ATCC 25922). While P. aeruginosa was insensitive to all compounds tested in disk diffusion method.
Inhibition effect of flavonoids on: E. coli
Inhibitory effect of polyphenols on invasion of VERO cells by E. coli
E. coli K12 strain was transformed with two plasmids: pGB2Ω inv/hly coding invasin a protein from Y. pseudotuberculosis, which allows penetration of bacteria into eukaryotic cells containing β-integrine in cell membrane and pAT505 coding GFP. Transformed E. coli were inoculated into VERO cells cultured in DMEM supplemented with 0–0.5 mM of analyzed compounds. After incubation, the cells were observed under optical microscopy to visualize bacteria containing green fluorescence protein, which penetrated VERO cells. Compounds 11 and 24 in 100 μM and 3 and 34 in 500 μM concentration prevent invasion of VERO cells by E. coli (Fig. 3c, d). However, 24 and 34 causes visible differences in morphology and amount of VERO cells.
Cytotoxicity analysis
The cytotoxic effect of polyphenols was determined using human HEK293 cells (Fig. 3e, f). The IC50 values were generally higher than MIC50, which suggest that tested compounds are either not toxic (pelargonidin) or only moderately toxic for human cells (Table 3).
Discussion
Growth of microbial resistance to antibiotics observed in recent years, becomes a serious challenge for contemporary medicine. 28 Responding to the urgent need to develop new effective treatments for bacterial infections, several new molecular targets were proposed. These are aminoacyl-tRNA synthetases, the important enzymes for cell life. 29 Their inhibitors are good candidates for therapeutics as shown previously.1,2,5,7,12,13 Differences in three-dimensional structure between eukaryotic and prokaryotic enzymes create a chance to obtain inhibitors specific only for bacterial proteins, acting as a potential antibacterial agent. As a therapeutic target we chose Tyr-RS due to significant changes in the structure between bacterial enzyme and their eukaryotic counterparts.29,30 Potential inhibitors were searched from natural compounds, mainly polyphenols. Selection of these groups of compounds was done based on preliminary results of docking various different compounds to TyrRS. The first screen of our collection of natural compounds containing polyphenols, alkaloids, and terpenes to isolate inhibitors of TyrRS was made based on in vitro tRNATyr aminoacylation with a mix of aaRSs isolated from S. aureus, P. aeruginosa, E. coli, and S. scrofa domestica (Table 2). Compounds showing low IC50 value were analyzed further using purified E. coli TyrRS to determine their Ki and mode of inhibition. KM and Vmax of E. coli TyrRS used in our experiments is in good agreement with the literature value. 31 Results of kinetic analysis of inhibition of aminoacylation presented by Lineweaver–Burk plot demonstrate that most of the compounds acts as competitive inhibitors. The inhibition constant (Ki −5.12 μM) of the most active inhibitor 11, was in the range of the KM −4.1 μM for TyrRS (Fig. 1b–g).
The mammalian cytoplasmic TyrRS was not inhibited in the analyzed range of inhibitor concentrations, which creates opportunity to use the flavonoids in prevention and treatment of bacterial infection. Antibacterial activities of TyrRS inhibitors examined in the present study were assessed by the presence of inhibition zone and minimal inhibitory concentration values. The compounds display varied activities depending on pathogen. Zone inhibition was observed only in the case of 31 and 36 for S. aureus and E. coli (Fig. 3a, b). Poor results in the case of flavonoids were probably due to their weak diffusion in agar. As can be seen from Table 3, MIC50 value for flavonoids were 30–480 μg/ml for Gram(+) bacteria and 80–425 μg/ml for Gram(−) bacteria. Values below 50 μg/ml were observed for 36, 1, 11, and 15 in the case of S. aureus and 1 and 35 in the case of P. aeruginosa. The most active against E. coli was 8.
Antibacterial activity of 1, 11, 24, and 34 was also checked by their effect on the infection of Vero cells by invasive E. coli strain LMG pGB2Ω inv/hly/pAT505. The presence of bacteria inside the cells was confirmed by observation of GFP fluorescence (Fig. 3c, d). The results showed that analyzed compounds effectively protected eukaryotic cell from pathogenic infection.
Despite the fact that phytochemicals, like flavonoids, terpenes, or alkaloids, are widely distributed in plants, regular diet cannot provide their higher amount. However, increasing of their concentration in plasma by supplementation is relatively easy. To confirm or exclude the obtained results further experiments on animals are necessary, particularly in the context of possible supplementation for preventive purposes.32–34 Even if the risk of pathological consequences of mutation incurred by the consumption of flavonoids was estimated as low, 35 it should be noted that a significant mutagenic effect has been detected in the case of some flavonoids, as well as in many other components of common food products. Additional disadvantage in the case of flavonoids is the fact that they inhibit a perplexing number and variety of eukaryotic enzymes and have a tremendously wide range of activities. In the case of enzyme inhibition, this has been postulated to be due to the interaction of enzymes with different parts of the flavonoid molecule.15,18,36
Structure–activity relationship (SAR) for different inhibitors from flavonoid group shows that flavones having more than two hydroxyl group at A ring, for example, 14 as well as more than one OH at B ring, for example, 15 are characterized by higher value of Ki compared with 13. However, removing of all OH groups from B ring as seen in 12 resulted in further decreasing of Ki, the best results, however, were observed in compound having methoxy group at 4′ position, for example, 11 (Tables 1 and 2). The same was observed also in a range of flavanols having 3-OH. There is almost no difference in Ki between 12, 13, and 2, 3. However, in the case of 1, the presence of 3-OH group, similar to the presence of B-ring at position 3 characteristic for isoflavones, for example, 24 results in the increase of Ki as compared with 11. Extending the molecule with sugar moiety at the benzopyran ring results in weakening the inhibitory potency of compounds.
Changes in molecule geometry through saturation of C2–C3 double bond also result in increasing of Ki as compared with 13 and 21, similar to increasing of the degree of freedom of molecule through breaking the C1–C2 bond as seen in 27. Structure comparison of the aminoacylation inhibitors shows that the most effective are those possessing the hydrophobic group at position 4′ and hydroxyl group at position 5 and 7.
Crystal structure of TyrRS in complex with 6 (fisetin) shows that it occupies the binding site approximately the same position as the tyrosyl moiety in the structure of human TyrRS. 37 The 7-OH group of the fisetin interact through hydrogen bonds with Tyr36 and Asp170, whereas ring B extends into the site binding the ribose of tyrosyladenylate and its 3′ and 4′-OH groups occupying approximately the positions O2′ and O3′ of the ribose. The remaining 3-OH substituents of fisetin project into the solvent region and make no interactions with the protein. 37 Although fisetin is not the most active inhibitor compared with 1 and 11, it binds in active site of TyrRS showing the competitive mode of inhibition (Fig. 4a–d).
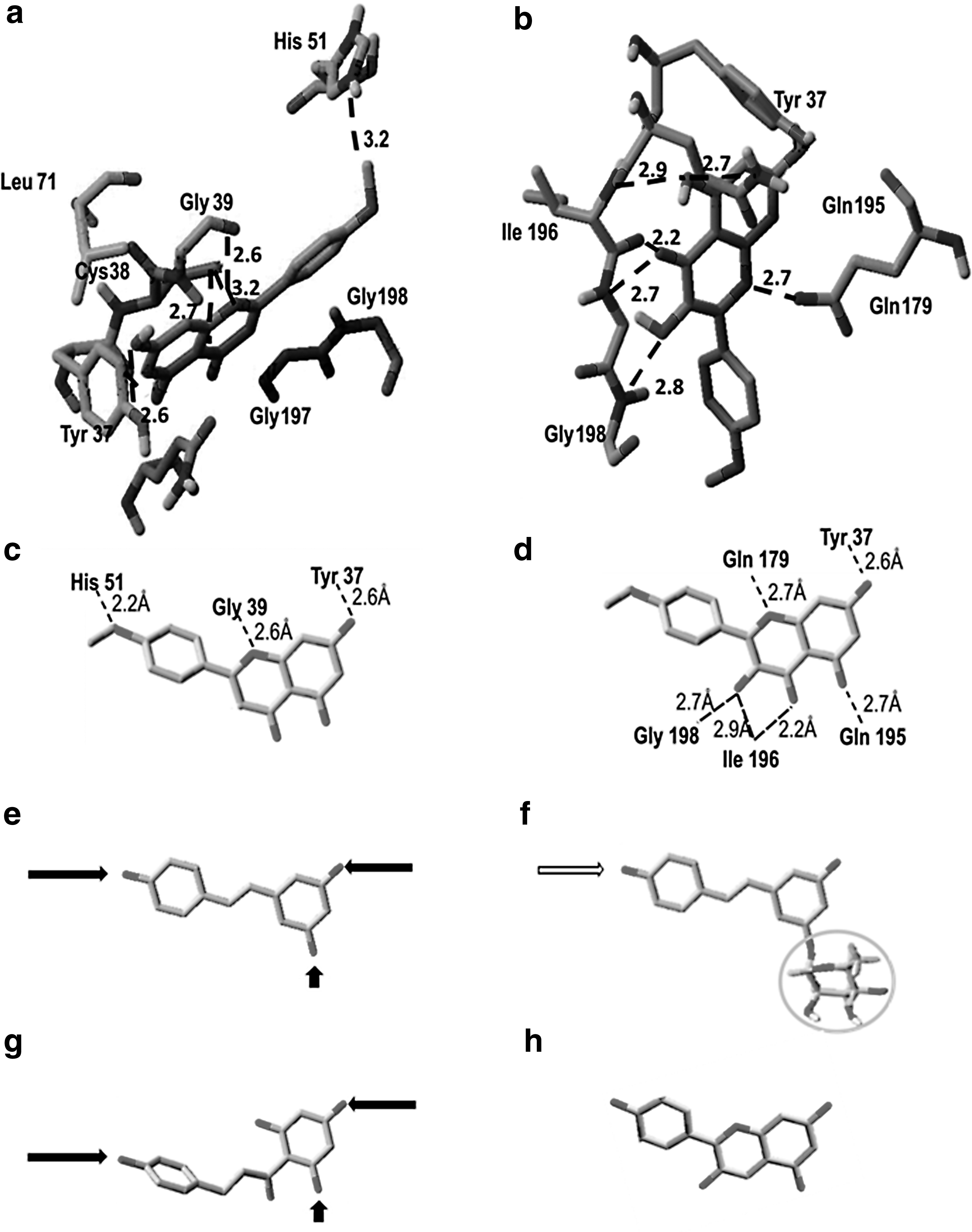
Interaction of acacetin (
To understand the good inhibitory activity of 1 and 11 observed experimentally, molecular docking of these ligands to binding site of TyrRS from E. coli was performed using the TyrRS complex structure. 13 The binding models of compounds 1 and 11 with TyrRS (Fig. 4a–d) show that due to the presence of OH group at position 5, both in 1 and 11, may form more hydrogen bonds with protein than 6, which may explain lower value of Ki.
Comparing the structure of 6 in complex with TyrRS with flavonoids from other groups, 27 and 29 show molecular similarity that allows them to interact in a competitive mode (Fig. 4e, h). However, differences in structure may explain the increasing value of Ki for these compounds. However, 29 (resveratrol) and 27 (phloretin), from structural point of view may be considered interacting with Tyr 37 through 5-OH of ring A as well as 4′-OH of ring B (Fig. 4e, h). However, substitution of 29 with sugar moiety at position 5 leading to formation of 30 (polydatin) affects the molecule geometry, which results in the decreasing of Ki and changes the mode of inhibition from competitive to mixed (Table 2 and Fig. 4f). Moreover, in the case of 28 and 30, in the presence of low concentration of inhibitors, the aminoacylation reaction was activated followed by the inhibition at higher concentration (Table 2 and Fig. 2). It may suggest existence of two different binding sites in the structure of TyrRS for these compounds. At lower concentration, flavonoid glycoside binds to higher affinity site, which results in enhancing of enzyme activity, and then interacts with lower affinity site, which leads to inhibition. Kinetic data show that 30 act as mixed inhibitor with higher affinity to Tyr-AMP-TyrRS complex. Polyphenolic compounds act usually as inhibitors, but a few examples of enzyme activation was also observed. 15 Different binding sites for low molecular inhibitor α-naphthoflavone were identified in case of CYP3A6, which resulted in stimulation or inhibition of its activities for specific substrates. 38 The only one compound among analyzed flavonoids, which inhibits TyrRS in uncompetitive mode is 34 (pelargonidin) (Table 2 and Fig. 4i). This may be attributed to the presence of a positive charge on the C ring, which interacts with protein negative charges. 39
In another case, substitution of aglycone at position 3 with sugar moiety weakens the inhibitory potency as seen for 8 (IC50 503 μM) or abolish as in the case of 9, 10, or 19 (Fig. 5) While 16 having glucose ring at position 7 of ring A still remains quite a potent inhibitor (IC50 118 μM), 19 substituted at position 8 (IC50 > 2,000 μM) does not inhibit TyrRS at all. Extending of aglycone with sugar moiety results in changing of inhibition mode toward noncompetitive, which means binding outside the active site.
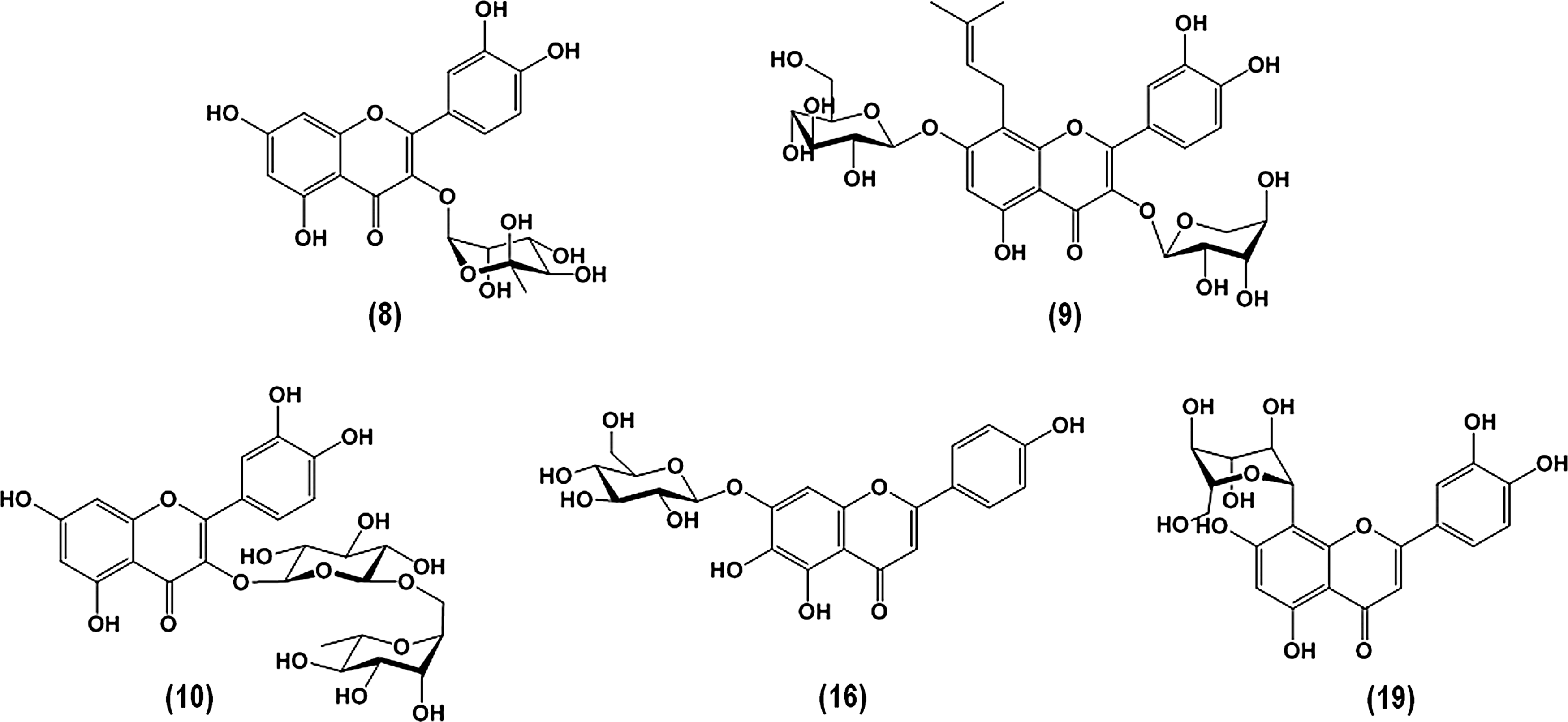
Structure of aglycones substituted with sugar moiety at different positions.
Another group of tested compound were alkaloids and terpenes, the most potent inhibitors of these groups are 35 (ursolic acid) and 36 (sanguinarine), which due to their structure act in a noncompetitive way. Other tested compounds, like 36, 37, 40, or 41 do not inhibit bacterial TyrRS.
The physiological tyrosine concentration in human blood amounts ca. 54 μM. 40 To ensure effective inhibition, concentration of tyrosine synthase inhibitor must be at least 100 times higher. However, flavonoids are not toxic to human cells and can be used in a higher concentration than tyrosine tRNA synthetase. Therefore, we suggest their application as dietary supplements and nutraceuticals as well as cosmeceuticals in the cosmetic industry.
Conclusions
In this study, we show results of screening of a series of polyphenolic compounds, terpenes, and alkaloids for their inhibitory activity against E. coli, S. aureus, and P. aeruginosa TyrRS. The most active competitive inhibitors of TyrRS are acacetin and kaempferide (Ki 5.1 and 9.1 μM, respectively). Among the compounds inhibiting in a noncompetitive way are epigallocatechin gallate, sanguinarine, and ursolic acid (Ki 5, 49, and 85 μM, respectively). Some of them inhibit S. scrofa domestica TyrRS, which is homologous to the human enzyme. These compounds inhibit the growth of E. coli and S. aureus and less P. aeruginosa strains.
SAR analysis shows that the most active flavonoid inhibitor should possess the hydroxyl group at position 5 and 7, as well as the hydrophobic group at 4′. These results were confirmed by molecular docking analysis.
Footnotes
Acknowledgments
This research was cofinanced by the Ministry of Science and Higher Education under the grants no. N N401 037236, N N405 126640, and by the National Science Center under the grant no. 2012/05/N/NZ7/02157.
Disclosure Statement
No competing financial interests exist.