
Abstract
Background:
Nontuberculous mycobacteria are habitants of environment, especially in aquatic systems. Some of them cause problems in immunodeficient patients. Over the last decade, 16S rRNA gene sequencing was established in 45 novel species of nontuberculous mycobacteria. Experiences revealed that this method underestimates the diversity, but does not distinguish between some of mycobacterium subsp. To recognize emerging rapidly growing mycobacteria and identify their subsp, rpoB gene sequencing has been developed.
Objectives:
To better understand the transmission of nontuberculous mycobacterial species from drinking water and preventing the spread of illness with these bacteria, the aim of this study was to detect the presence of bacteria by PCR-sequencing techniques.
Materials and Methods:
Drinking water samples were collected from different areas of Kermanshah city in west of IRAN. After decontamination with cetylpyridinium chloride, samples were filtered with 0.45-micron filters, the filter transferred directly on growth medium waiting to appear in colonies, then DNA extraction and PCR were performed, and products were sent to sequencing.
Results:
We found 35/110 (32%) nontuberculous mycobacterial species in drinking water samples, isolates included Mycobacterium goodii, Mycobacterium aurum, and Mycobacterium gastri with the most abundance (11.5%), followed by Mycobacterium smegmatis, Mycobacterium porcinum, Mycobacterium peregrinum, Mycobacterium mucogenicum, and Mycobacterium chelonae (8%).
Conclusions:
In this study, we recognized the evidence of contamination by nontuberculous mycobacteria in corroded water pipes. As a result of the high prevalence of these bacteria in drinking water in Kermanshah, this is important evidence of transmission through drinking water. This finding can also help public health policy makers control these isolates in drinking water supplies in Kermanshah.
Background
N
Although person-to-person transmission occurs rarely, these organisms can produce serious morbidity because of hydrophobicity. The fatty acid-rich cell wall is responsible for virulence and the ability to survive in harsh environmental conditions such as limitation of food and oxygen. They can tolerate different pH ranges, temperatures, and chlorine in water, making them potentially laborious to eliminate. 4 A recent study using DNA fingerprint analysis revealed that household water was the source of mycobacterial infection in patients with NTM disease. According to other studies, Mycobacterium avium subsp avium, Mycobacterium intracellulare, Mycobacterium goodii, Mycobacterium smegmatis, Mycobacterium mucogenicum, Mycobacterium chelonae, Mycobacterium lentiflavum, and Mycobacterium gordonae are among the rapid and slow growing NTM existing in water supplies. 5 In past decades, mycobacteria were identified using phenotypic traits, such as colony morphology, growth rates, pigment production, and biochemical profiles. These methods are laborious to set up, and results are sometimes unreliable and often not reported in a timely manner to guide clinical decisions because different species (especially newly defined ones) may have indistinguishable morphological and biochemical profiles. Therefore, recently developed molecular methods have facilitated the rapid and reliable identification of many mycobacterial species. Nucleic acid probes, species-specific PCR, reverse hybridization, and 16S rRNA sequencing have been used in mycobacterial laboratories. 6
DNA-based molecular methods such as PCR-sequencing have high sensitivity and specificity and can detect small amounts of bacterial DNA samples faster than traditional methods. 16S rRNA and rpoB gene sequencing is a more discriminative identification technique that could improve clinical care of NTM disease with less turnaround time and more accuracy. In addition, this technique was used more for cases that have many interspecies, such as NTM. 7 Therefore, we aimed to assess the presence of NTM in drinking water distribution systems by analyzing samples from various parts of Kermanshah, west Iran. 4
Materials and Methods
Sample collection and culture
Sample size calculation was according to the criteria described by other investigators. 8 Because of limitations in time and facilities in our laboratory, providing this sample size was impossible. So, we prepared samples according to the Kermanshah water and sewage map, which was divided into 11 regions.
A total of 110 samples were collected from 11 well-serviced urban areas. For water and tap water sampling, Kermanshah city was divided into 11 regions, and in each region, 4 samples were taken from water sources and 7 samples from tap water. Samples were collected during the period of June to September 2015. Before sample collection, water was allowed to run to waste at a uniform rate for 2–3 min. At the sampling location, pH and temperature were measured and the total chlorine content was determined using the DPD method (N,N-diethyl-p-phenylenediamine colorimetric method). The volume of water samples was 0.5 or 1 L in most studies about drinking water; because of more samples and less time and according to the volume of filtration set at 0.25 L, we collected water samples of 0.5 L volume in sterile glass bottles. We added sodium thiosulfate to a final concentration of 0.01% (wt/vol) to neutralize any free or combined residual chlorine. Cetylpyridinium chloride 0.01% (wt/vol) was added as an antimicrobial agent to eliminate nontarget microorganisms, a substance recommended by most studies for the isolation of mycobacteria from drinking water with low contamination rates and high isolation.4–9 All samples were transferred to the laboratory on ice and analyzed. Water samples were filtered through 0.45-μm pore size nitrocellulose filters (Millipore). 4
Filters were transferred directly onto Lowenstein–Jensen (LJ) slant media and incubated at 37°C. The plates were examined once a week for 8 weeks for colony morphology, growth rate, and pigmentation. When the colonies appeared, they were exposed to Ziehl–Neelsen acid-fast staining. 9
DNA extraction
Briefly, 2–3 loopful of bacterial growth from each isolate was transferred to an Eppendorf tube containing 200 μl of sterile distilled water. The suspension was boiled in a water bath for 10 min. Then, the suspension was vortexed for 10 sec and centrifuged at 14,000 rpm for 15 min. The clear supernatant containing DNA was transferred to a new microtube and stored at −20°C.
To evaluate the purity of the extracted DNA, a spectrophotometer (260/280 ≥ 1.7 OD) was used. The quality of extracted DNA was controlled on agarose gel 2%. 10
Species identification of NTM by PCR-sequencing
PCR was performed for 750 bp rpoB and 1,500 bp 16S rRNA genes. 16S rRNA gene primers were 5′-AGAGTTTGATCCTGGCTCAG-3′ and as reverse: 5′-TACCTTGTTACGACTT-3′ and rpoB gene primers were MycoF 5′-GGCAAGGTCACCCCGAAGGG-3′ and MycoR 5′-AGCGGCTGCTGGGTGATCATC-3′. 11
Amplification was done in a thermal cycle C1000 (BioRad) under the following conditions. PCR assays were performed in a final volume of 50 μl, containing 5 μl DNA template, 5 μl of 10× buffer, 2 mM MgCl2, 0.2 mM dNTP, 0.4 mM each primer, and 2 U Taq polymerase. 10 The reaction cycle was 30-fold and included primary heating to 94°C for 5 min, cycles of denaturation (94°C, 1 min), annealing (55°C, 1 min), and extension (72°C, 5 min), followed by a final extension (72°C, 5 min). Expected lengths of the amplicons were ascertained by electrophoresis in agarose gel 1%. Bands were visualized in a Gel Doc system (BioRad) under ultraviolet light after ethidium bromide staining. 11
PCR-sequencing analysis
The purified PCR products from the water samples and reference mycobacterial strains were delivered to Bioneer Company for sequencing. A phylogenetic tree of the 35 NTM was obtained from 16S rRNA and rpoB sequences using the neighbor-joining method with Kimura's two-parameter (K2P) distance correction model with 1,000 bootstrap replications in the MEGA version 7 software package. The tree was rooted using Mycobacterium tuberculosis. The sequences determined in this study have been deposited in GenBank/EMBL/DDBJ. 12
Nucleotide sequence accession numbers:
The GenBank accession numbers of investigated isolates of NTM determined in this study for the rpoB gene and 16S rRNA include KU861812, KU861813, KU861814, KU861815, KU861816, KU861817, KU861818, KU861819, KU861820, KU861821, KU861822, KU861823, KU861824, KU861825, KU861826, KU861827, KU861828, KU861829, KU861830, KU861831, KU861832, KU861833, KU861834, KU861835, KU861836, KU861837, KU861838, KU861839, KU861840, KU861841, KU861842, KU861843, KU861844, KU861845, KU861846, KU861847, KU861848, KU861849, KU861850, KU861851, KU861852, KU861853, KU861854, KU861855, KU861856, KU861857, KU861858, KU861859, KU861860, KU861861, KU861862, KU861863, KU861864, KU861865, KU861866, KU861867, KU861868, KU861869, KU861870, KU861871, KU861872, KU861873, KU861874, KU861875, KU861876, KU861877, KU861878, KU861879, KU861880, and KU861881.
Results
Thirty-five (32%) isolates from 110 samples were classified as Mycobacterium species by performing culture and acid-fast staining. The average range of chlorine, pH, and temperature was 0.15 ppm, 7.5, and 21°C, respectively.
All 35 isolates belonged to 14 different species with the sequencing of the rpoB gene and underwent additional 16S rRNA gene sequencing. rpoB gene sequencing per se identified 14/14 (100%) of species as Mycobacterium species, whereas 16S rRNA gene sequencing identified only 11/14 (78%) of the species to be Mycobacterium species. The three species not detected through 16S rRNA gene sequencing were Mycobacterium massiliense, M. gordonae, and M. avium subspecies avium (Figs. 1 and 2). Therefore, rpoB gene sequencing can be a preferred method for detecting NTM intraspecies.
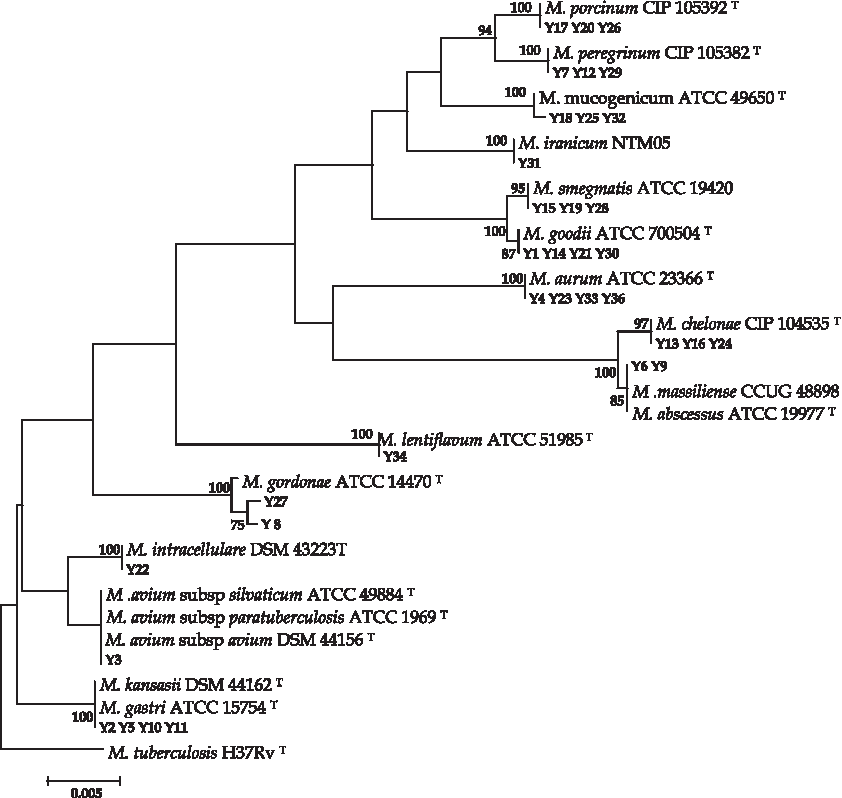
Phylogenetic tree of 16S rRNA gene sequences of mycobacterial species by using the neighbor-joining method and Kimura's two-parameter distance correction model. The support of each branch, as determined from 1,000 bootstrap samples, is indicated by the value at each node (as a percentage). Mycobacterium tuberculosis was used as outgroup. Bar, 0.5% difference in nucleotide sequences.

Phylogenetic tree of rpoB gene sequences of mycobacterial species by using the neighbor-joining method and Kimura's two-parameter distance correction model. The support of each branch, as determined from 1,000 bootstrap samples, is indicated by the value at each node (as a percentage). Mycobacterium tuberculosis was used as outgroup. Bar, 2% difference in nucleotide sequences.
As shown in Table 1, M. goodii, Mycobacterium aurum, and Mycobacterium gastri were the most prevalent species (11.5%), followed by M. smegmatis, Mycobacterium porcinum, Mycobacterium peregrinum, M. mucogenicum, and M. chelonae with a prevalence of 8%, and M. gordonae and M. massiliense with a prevalence of 5%.
The pollution frequency reported in our study (30%) compared with other studies (10–24.7% in Iran and up to 54% in other areas) is quite considerable. This amount of pollution in the pipes is probably due to the old water distribution system in Kermanshah with a lifetime of 40 years, which increases the likelihood of biofilm formation in the water distribution system.
Discussion
NTM are widely distributed in the environment, soil, water, and other natural reservoirs. In recent years, NTM have emerged as a major cause of opportunistic infections. Therefore, it is necessary to perform risk analysis and identify the species of NTM that are present in the environment for understanding their incidence and informing clinicians. 4 It is noteworthy that human NTM illness is more frequently reported during the summer and autumn. 1
In our study, the dominant species of mycobacteria isolated from water samples included M. goodii, M. gastri, and M. aurum (11%), followed by M. smegmatis, M. porcinum, M. peregrinum, M. mucogenicum, and M. chelonae (8%). M. goodii is associated with sporadic cases of cellulitis, osteomyelitis, infected pacemaker sites, lipoid pneumonia, and bursitis and develops wound infections after surgery. 13 M. aurum cause keratitis and catheter-related bacteremia and bacteremia in immunocompromised children, as well as bilateral pneumonia in patients receiving infliximab therapy. M. aurum infection is a potential threat for managing critical cases of ocular TB even in patients with no predisposing factors. 14
M. gastri is involved in chronic obstructive pulmonary disease, with chronic respiratory failure. It is also related to pneumonia in immunocompromised patients and disseminated disease in children. 15 M. mucogenicum develops bloodstream infections in patients with sickle cell disease in an outpatient setting. Pulmonary disease due to M. mucogenicum has been reported in the TB Registry of the Czech Republic. 15 M. chelonae is probably the most common NTM responsible for skin and soft tissue infections. 16 In one study on water samples collected from the faucet, NTM species were recovered from 16% of the samples and included M. mucogenicum, M. porcinum, M. avium, M. gordonae, Mycobacterium cosmeticum, Mycobacterium fortuitum, and Mycobacterium sp. 1 In three different studies in Isfahan, Iran, the researchers found that M. chelonae, M. mucogenicum, and M. gordonae 17 and M. smegmatis, M. mucogenicum, M. gordonae, and M. avium ssp. 18 were dominant isolates with NTM prevalence of 13.3%, 24%, and 36%, 19 respectively.
Castillo-Rodal identified M. peregrinum, M. smegmatis, M. gordonae, and M. avium ssp. Hominissuis from three Mexican aquatic systems. All of these species have been previously isolated from different water sources. 5 Covert isolated different species of NTM from 54% of ice samples and 35% of public drinking water sources. 20
Carson found that M. chelonae isolates were able to multiply in distilled water, attaining population levels of 105–106 cells/ml. Results of disinfectant studies showed that sterile distilled water-grown cells of M. chelonae were markedly resistant to chlorine. M. smegmatis persists in treated effluents and is 20–100 times more resistant than coliforms and other gram-negative bacteria to free chlorine residuals up to 1.0 μg/ml. Moreover, M. aurum and M. gordonae were still 100 and 330 times more resistant to chlorine than Escherichia coli, respectively. 21 Shojaei found a novel species called Mycobacterium iranicum sp. Nov. isolated in Iran as one of the first detected strains from clinical samples, 22 but we identified it for the first time from drinking water systems.
rpoB gene sequencing per se identified 14/14 (100%) of species as Mycobacterium species, whereas 16S rRNA gene sequencing identified only 11/14 (78%) of the species to be Mycobacterium species. The three species not detected through 16S rRNA gene sequencing were M. massiliense, M. gordonae, and M. avium subspecies avium. 16S rRNA gene sequencing has been used as the reference method for identifying unusual mycobacterial isolates. This approach has contributed to the description of 45 novel NTM species over the last 10 years. However, the investigation of large NTM collections by 16S rRNA gene sequencing has not resolved the classification of all isolates. Indeed, 16S rRNA gene sequence identity alone is not a useful method for the identification of mycobacteria because the intraspecies similarity of the 16S rRNA gene ranges from 94% to 100%; therefore, it cannot distinguish between closely related species with identical 16S rRNA gene sequences, and ambiguous results have been found. rpoB gene sequencing has been proposed for the classification of closely related isolates that were misidentified by 16S rRNA gene sequencing.11,23 In our study, the average range of chlorine, pH, and temperature was 0.15 ppm, 7.5, and 21°C, respectively. The results of studies in Iran and other areas indicate a correlation between the mycobacterial presence in water samples and temperature. Temperature affects microbial propagation in drinking water systems by increasing growth and indirectly by reducing the effect of chlorine. So, bacterial counts were four and two times higher at 20°C than at 7°C, respectively, showing that temperature is a significant factor in bacterial proliferation in drinking water. 24 In our study, the prevalence of Mycobacterium species was 32%, which can be attributed to the high range of temperature. As previously reported, 21 NTM are resistant to chlorine residues added to water supplies, and analyses do not show a significant relationship between NTM proliferation and chlorine concentrations.
Conclusion
We found that rpoB-based sequencing alone is a preferred procedure for identifying NTM species in water samples compared with 16S rRNA sequencing. Our drinking water is polluted with NTM, especially with M. goodii, M. gastri, and M. aurum that are related to diseases such as bacteremia, cellulitis, osteomyelitis, infected pacemaker sites, and lipoid pneumonia. We can state that Kermanshah drinking water is potentially polluted and can cause severe problems for immunocompromised patients, people receiving chemotherapy, and also create nosocomial infections in hospital water supplies. This finding can also help public health policymakers control these isolates in drinking water supplies in Kermanshah.
Footnotes
Acknowledgments
The data were obtained from an MSc thesis in microbiology by L.Y. The study was financially supported by the Kermanshah University of Medical Sciences. The authors thank members of the department of microbiology in Kermanshah University of Medical Sciences, particularly for their assistance with the PCR assessment in this study. The study was financially supported by the Kermanshah University of Medical Sciences grant 93361.
Author Contributions
All authors had an equal role in design, work, and performing the laboratory works.
Disclosure Statement
No competing financial interests exist.