
Abstract
Novel therapeutic approaches are urgently needed to combat nosocomial infections caused by extremely drug-resistant (XDR) “superbugs.” This study aimed to investigate the synergistic antibacterial activity of polymyxin B in combination with selective estrogen receptor modulators (SERMs) against problematic Gram-negative pathogens. In vitro synergistic antibacterial activity of polymyxin B and the SERMs tamoxifen, raloxifene, and toremifene was assessed using the microdilution checkerboard and static time–kill assays against a panel of Gram-negative isolates. Polymyxin B and the SERMs were ineffective when used as monotherapy against polymyxin-resistant minimum inhibitory concentration ([MIC] ≥8 mg/L) Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter baumannii. However, when used in combination, clinically relevant concentrations of polymyxin B and SERMs displayed synergistic killing against the polymyxin-resistant P. aeruginosa, K. pneumoniae, and A. baumannii isolates as demonstrated by a ≥2–3 log10 decrease in bacterial count (CFU/ml) after 24 hours. The combination of polymyxin B with toremifene demonstrated very potent antibacterial activity against P. aeruginosa biofilms in an artificial sputum media assay. Moreover, polymyxin B combined with toremifene synergistically induced cytosolic green fluorescence protein release, cytoplasmic membrane depolarization, permeabilizing activity in a nitrocefin assay, and an increase of cellular reactive oxygen species from P. aeruginosa cells. In addition, scanning and transmission electron micrographs showed that polymyxin B in combination with toremifene causes distinctive damage to the outer membrane of P. aeruginosa cells, compared with treatments with each compound per se. In conclusion, the combination of polymyxin B and SERMs illustrated a synergistic activity against XDR Gram-negative pathogens, including highly polymyxin-resistant P. aeruginosa isolates, and represents a novel combination therapy strategy for the treatment of infections because of problematic XDR Gram-negative pathogens.
Introduction
T
New antibiotic therapeutic strategies are urgently needed to treat infections caused by bacterial “superbugs,” in particular Gram-negative Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae. A novel, but rational, approach is repurposing FDA-approved drugs for alternative indications such as antimicrobial agents.4,5
Polymyxin B and colistin (polymyxin E) are lipopeptide antibiotics indicated as a last-line treatment for XDR Gram-negative bacterial infections (Fig. 1). 6 Worryingly, reports of polymyxin-resistant isolates are becoming more and more common, including very recent Enterobacteriaceae isolates with plasmid-carrying mcr-1.7–9 Therefore, novel strategies to maintain the efficacy of these important last-line antibiotics against problematic XDR pathogens are crucial. An innovative strategy that is gaining momentum is the synergistic use of antibiotics with FDA-approved nonantibiotics. 10 In this novel approach, an FDA-approved nonantibiotic drug is combined with a specific antibiotic that enables it to exert its “off-target” antibacterial properties against the bacterial cell.

Structures of the SERMs toremifene, tamoxifen, and raloxifene, and the lipopeptide antibiotic polymyxin B1. The cationic side chains of the five diaminobutyric acid (Dab) residues of polymyxin B are highlighted by darker gray circles and the hydrophobic segments are highlighted by lighter gray. SERMs, selective estrogen receptor modulators.
Selective estrogen receptor modulators (SERMs) tamoxifen, raloxifene, and toremifene are currently used for the treatment of advanced breast cancer and osteoporosis in premenopausal and postmenopausal women (Fig. 1).11–14 Notably, SERMs have been previously reported to display direct antifungal, antiviral, and antiparasitic activities, making them perfect candidates for nonantibiotic–antibiotic synergy testing.15–19 The purpose of this study was to investigate the in vitro synergistic antibacterial activity and mechanistic studies of SERMs in combination with polymyxin B against problematic XDR Gram-negative pathogens, including those resistant to polymyxins.
Methods
Materials
All compounds including polymyxin B (Catalogue No. 81334; ≥6,500 IU/mg) were purchased from Sigma-Aldrich (Melbourne, Australia). Polymyxin nonapeptide was produced through enzymatic cleavage of the fatty acyl diaminobutyric acid (Dab) 1 from polymyxin E and HPLC purified as previously described.20,21
Bacterial isolates
All bacterial strains used in this study are described in Table 1, including 13 mucoid and nonmucoid clinical isolates of P. aeruginosa selected from patients with acute exacerbations of cystic fibrosis. 22 A reference strain, P. aeruginosa ATCC 27853 (polymyxin B minimum inhibitory concentration [MIC] = 1 mg/L) (American Type Culture Collection, Rockville, MD), was examined. Resistance to polymyxin B was defined as MICs of ≥8 mg/L. 23 Four different K. pneumoniae isolates including polymyxin-susceptible and polymyxin-resistant isolates were also studied, including a reference strain K. pneumoniae ATCC 13883. A total of eight A. baumannii strains were examined in this study, including four polymyxin-susceptible and four polymyxin-resistant isolates.
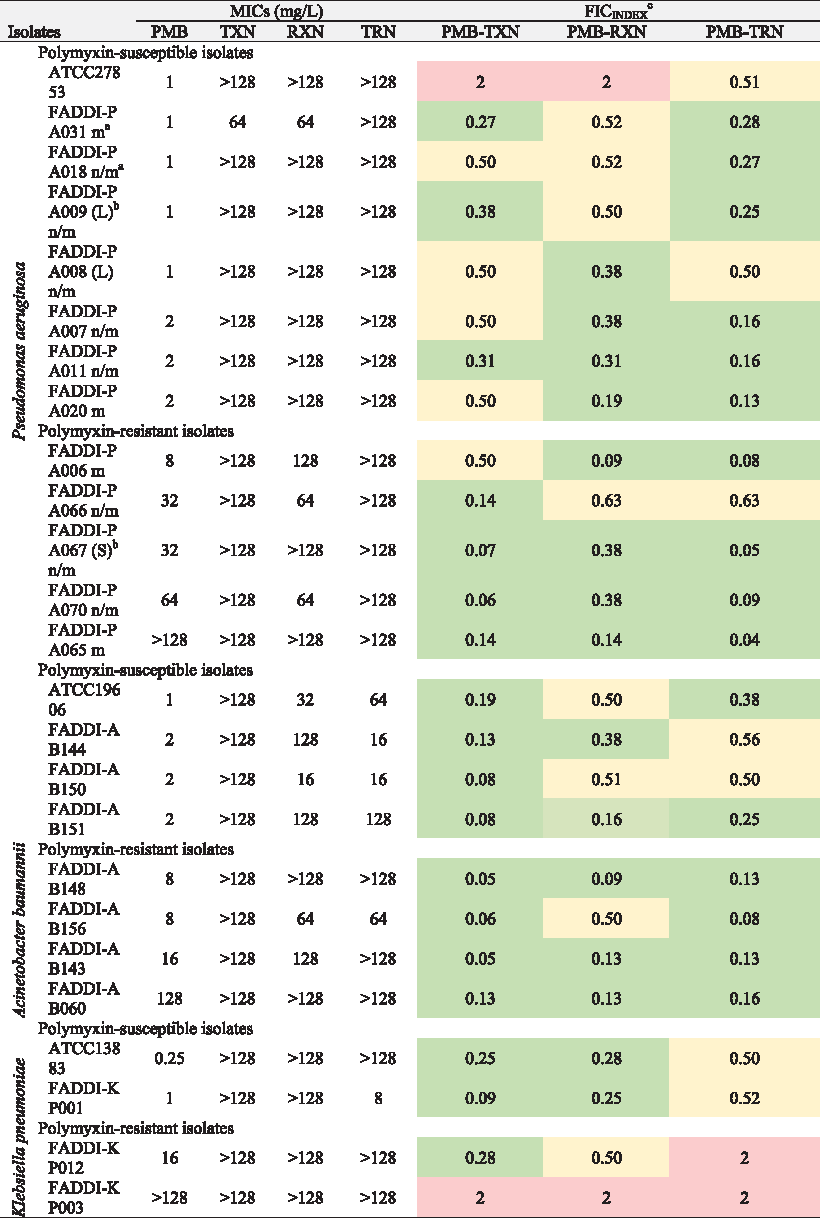
n/m, nonmucoid; m, mucoid.
L, large colony; S, small colony.
FIC = (MIC of polymyxin B in combination/MIC polymyxin B alone) + (MIC antineoplastic drug in combination/MIC antineoplastic alone); synergism FIC <0.5 [green]; addition FIC = 0.5–1.0 [yellow]; indifference FIC = 1–4 [red]; antagonism FIC ≥4 [not observed].
In vitro antibacterial activity
MICs were performed according to the Clinical and Laboratory Standards Institute guidelines. 24 MICs were determined for all isolates in three replicates on separate days using broth microdilution method in cation-adjusted Mueller–Hinton broth (CAMHB). Fractional inhibitory concentrations (FICs) were determined using the microdilution checkerboard method (FICINDEX values were interpreted as follows: synergism ≤0.5; additive = 1, indifferent >1 but <2 and antagonism ≥2). 25 If no endpoint MIC value could be determined because of the combination of resistance and the insolubility of the SERM compounds, the next highest MIC value was chosen for the FIC calculation (i.e., if the MIC >128, an MIC = 256 was selected for the calculation). Therefore, the reported FIC values are essentially conservative estimates; in effect if it was possible to determine the endpoint MIC, then the FIC values would be lower (i.e., better synergy). The static time–kill kinetics of polymyxin B per se and in combination with SERMs were investigated against select Gram-negative isolates, selected based on the determined FIC values. 26 Aliquots (∼200 μl) of early log-phase bacterial suspensions were inoculated into 50 ml polypropylene tubes (Greiner Bio-one, Frickenhausen, Germany) containing 20 ml of CAMHB containing DMSO (2.5% v/v) and drug at the specified concentrations. Colonies were counted by a ProtoCOL-automated colony counter (Synbiosis, Cambridge, United Kingdom) after incubation for 24 hours at 35°C. The lower limit of counting was 20 CFU/ml.
Electron microscopy
Scanning and transmission electron microscopies (SEM and TEM) were conducted as previously described in detail after treatment against P. aeruginosa FADDI-PA067 cells with polymyxin B alone and in combination with toremifene for 2 hours. 27 Confocal imaging of the released cytosolic green fluorescence protein (GFP) was performed using P. aeruginosa strain Pa AH298-GFP expressing GFP under the control of the growth rate-dependent rrnBp1 promoter that was kindly provided by Professor Philip S. Stewart. 28
Nitrocefin assays
The outer membrane (OM) permeabilizing activity of the drugs was assessed using the β-lactamase nitrocefin assay. 29 In brief, the β-lactamase positive P. aeruginosa strain FADDI-PA067 (P. aeruginosa strains were tested for β-lactamase production using nitrocefin disk; Sigma-Aldrich) was subcultured on a nutrient agar at 35°C overnight. The culture (derived from a single colony) was grown to exponential phase and harvested by centrifugation (10,000 g; 10 minutes), washed twice in PBS, pH 7.2, and resuspended at an OD600nm = 0.50. Nitrocefin (2 mM) was prepared in DMSO. The assay was performed in 96-well plates. The 200 μl reaction mixture consisted of 100 μl bacterial suspension, 96 μl drug (8 mg/L polymyxin B alone or in combination with 8 mg/L toremifene), and 4 μl of 2 mM nitrocefin solution. The absorbance (OD495nm) was measured at room temperature for 140 seconds.
Artificial sputum media biofilm assay
The artificial sputum media (ASM) assays were performed with P. aeruginosa strain FADDI-PA067 in a 24-well plate format. ASM culture was freshly prepared as described by Kirchner et al. and filter sterilized.29,30 P. aeruginosa strain FADDI-PA067 was subcultured on a nutrient agar and incubated at 35°C overnight. The overnight culture was diluted in CAMHB to an OD600nm 0.05 ± 0.01 and then further diluted 1:100 in fresh ASM. A total volume of 1.8 ml was added to each well and incubated for 3 days under aerobic condition at 37°C before the addition of polymyxin B and toremifene alone or in combination for 24 hours. Viability was determined, wherein 100 μl of 0.02% (v/v) resazurin was added to each well and incubated for 2 hours at 37°C. Fluorescence of each well was measured using an ENVISION plate reader (PerkinElmer, Australia) at excitation 530 nm and emission 590 nm. Fluorescence was corrected by the subtraction of background noise. Cell viability was calculated as (mean fluorescence of treated biofilms/mean fluorescence of untreated control) × 100%. For each antibiotic concentration, four technical replicates were conducted.
Detection of cellular reactive oxygen species production
The production of reactive oxygen species (ROS) in P. aeruginosa strain FADDI-PA067 after antibiotic treatment was detected using the CellROX Green dye (Lifetechnologies, Melbourne, Australia). Fluorescence was measured using a Cary Eclipse Fluorescence spectrophotometer (Varian, Mulgrave, Australia) at excitation 485 nm, emission spectrum 490–600 nm. Slit widths were set to 5 nm for both the excitation and emission monochromators.
Cytoplasmic membrane depolarization assay
Cytoplasmic membrane depolarization was determined using cyanine dye diSC3-5 (3,3-dipropylthiadicarbocyanine iodide). 31 The assay was performed as previously described with slight modification. 32 Mid-log phase culture (1 ml) was harvested by centrifugation at 4,000 g for 10 minutes at room temperature and resuspended in 1 ml of assay buffer (5 mM HEPES, 20 mM glucose, pH 7.4). The bacteria suspension was diluted 100-fold using the assay buffer to reach the OD600nm of 0.005. Cells were treated with 0.2 mM EDTA (pH 8.0) then with 0.4 μM diSC3-5 and incubated for 1 hour at room temperature. The uptake of diSC3-5 into bacterial cells was monitored using a Tecan Infinite® m1000 Pro Multi-mode reader. Then, 0.1 M KCl was added to equilibrate the cytoplasmic and external K+ concentrations and 90 μl of the cell suspension was transferred to a 96-well black-walled plate (Corning #3609). After the fluorescence level stabilized, 10 μl of a concentration series of compounds was added into each well and the fluorescence intensity (excitation/emission: 620/670) was measured every minute for 40 minutes. Vehicle solution (10 μl H2O) was used as a negative control in the assay.
Results
In vitro antimicrobial synergy of polymyxin B in combination with SERMs
Polymyxin B and the SERMs tamoxifen, raloxifene, and toremifene were screened for direct antimicrobial activity against a panel of polymyxin-susceptible and polymyxin-resistant clinical isolates of P. aeruginosa, K. pneumoniae, and A. baumannii (Table 1). The polymyxin B–SERM combinations displayed excellent synergy against the polymyxin-resistant isolates of each species (FICI 0.04–0.38). Except for the appreciable activity of toremifene against K. pneumoniae FADDI-KP001 (MIC 8 mg/L), the SERMs per se were not active against any of the P. aeruginosa, K. pneumoniae, and A. baumannii isolates (MICs 64 to >128 mg/L). The combination of polymyxin B with tamoxifen displayed synergistic activity against 8 of the 13 P. aeruginosa isolates, including all 5 polymyxin-resistant isolates, the 8 A. baumannii isolates, and 3 out of 4 K. pneumoniae isolates. The combination of polymyxin B with raloxifene displayed synergistic activity against eight P. aeruginosa isolates, five A. baumannii isolates, and two K. pneumoniae isolates. The combination of polymyxin B with toremifene displayed synergistic activity against 10 out of 13 P. aeruginosa isolates, 6 A. baumannii isolates, but none of the K. pneumoniae isolates. To probe whether the synergy between polymyxin B and the SERMs is a result of the permeabilizing activity of polymyxin on the Gram-negative OM that allows the SERMs to enter the cells and reach their intracellular targets, we tested polymyxin nonapeptide in combination with tamoxifen against two polymyxin-resistant P. aeruginosa strains (FADD-PA067 and FADDI-PA070) and the polymyxin-sensitive strain P. aeruginosa ATCC 278853. Polymyxin nonapeptide has the N-terminal fatty acyl Dab 1 segment removed and consequently lacks the direct bactericidal activity of the mature polymyxin molecule, although it retains the OM permeabilizing activity and is often employed as a sensitizer for other antibiotics. 21 The polymyxin nonapeptide–tamoxifen combination was inactive against P. aeruginosa strains FADD-PA067 and FADDI-PA070, whereas good synergy (FIC = 0.18) was observed against P. aeruginosa ATCC 278853.
Antibacterial killing kinetics of the polymyxin B–SERM combinations
The synergistic killing activity of the polymyxin B–SERM combinations in static time–kill studies was assessed against seven Gram-negative clinical isolates (Fig. 2). Clinically relevant concentrations of the drugs were used in the time–kill experiments.6,12–14 The antipseudomonal activity of the polymyxin B–SERM combinations was assessed against FADDI-PA070 (polymyxin B MIC 64 mg/L; tamoxifen >128 mg/L), FADDI-PA065 (polymyxin B MIC >128 mg/L; tamoxifen >128 mg/L), FADDI-PA067 (polymyxin B MIC 32 mg/L; toremifene >128 mg/L), and FADDI-PA006 (polymyxin B MIC 8 mg/L; raloxifene >128 mg/L). Monotherapy with polymyxin B or each SERM exhibited no antibacterial activity against the P. aeruginosa isolates up to 24 hours with the bacterial killing curves indistinguishable from that of the control.

Time–kill curves for polymyxin B–SERM combinations against polymyxin-susceptible and polymyxin-resistant clinical isolates of Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter baumannii.
The polymyxin B–tamoxifen combination displayed a ∼4 log10 CFU/ml decrease against P. aeruginosa FADDI-PA070 over 24 hours as compared with the control. The polymyxin B–tamoxifen combination displayed a ∼3.2 log10 CFU/ml decrease against P. aeruginosa FADDI-PA065 over 24 hours as compared with the control. The polymyxin B–toremifene combination displayed a decrease in P. aeruginosa FADDI-PA067 bacterial count of ∼5.7 log10 CFU/ml over 24 hours as compared with the control. The polymyxin B–raloxifene combination displayed a decrease in P. aeruginosa FADDI-PA006 bacterial count of ∼3.5 log10 CFU/ml over 24 hours as compared with the control.
The killing kinetics of polymyxin B–tamoxifen combinations against A. baumannii were assessed against a polymyxin-susceptible strain ATCC 19606 (polymyxin B MIC 1 mg/L; tamoxifen MIC >128 mg/L) and a polymyxin-resistant XDR clinical isolate FADDI-AB156 (ST22 containing OXA-23, OXA-66, TEM-116, PER-1, and ADC-29) 33 (polymyxin B MIC 8 mg/L; tamoxifen >128 MIC mg/L). The polymyxin B–tamoxifen combination performed well against A. baumannii ATCC 19606 and caused a decrease in bacterial count of ∼4.5 log10 CFU/ml over 24 hours as compared with the control. However, despite the good FICI (0.07), the polymyxin B–tamoxifen combination displayed moderate activity against the XDR polymyxin-resistant A. baumannii FADDI-AB156 isolate, with a decrease of ∼5 log10 CFU/ml over 2–8 hours; although extensive regrowth occurred at 24 hours.
Finally, we examined the killing kinetics of the polymyxin B–tamoxifen combination against the polymyxin-susceptible K. pneumoniae FADDI-KP001 isolate (polymyxin B MIC 1 mg/L; tamoxifen >128 MIC mg/L) and the polymyxin-resistant K. pneumoniae FADDI-KP012 isolate (polymyxin B MIC 16 mg/L; tamoxifen >128 MIC mg/L). Against K. pneumoniae FADDI-KP001, the polymyxin B–tamoxifen combination decreased the bacterial count ≥6 log10 CFU/ml over 1–2 hours, followed by extensive regrowth from 4 to 24 hours. Irrespective of the good FICI (0.26), the polymyxin B–tamoxifen combination displayed poor activity against K. pneumoniae FADDI-KP012, seen as a decrease in bacterial count of ∼3 log10 CFU/ml over 1–2 hours, followed by extensive regrowth from 4 to 24 hours.
Overall, the polymyxin B–toremifene combination displayed the best killing kinetics and as such was selected for the following antipseudomonal mechanistic studies. To assess whether the rebound growth seen at 24 hours relates to polymyxin B resistance, we recovered the cells from an isolate of each species at 24 hours and tested the polymyxin B MIC. In the case of K. pneumoniae FADDI-KP001 (polymyxin B MIC 1 mg/L), the polymyxin MIC of the rebound cells did not change significantly, 2 mg/L; for A. baumannii FADDI-AB156 (polymyxin B MIC 8 mg/L), the polymyxin MIC of the rebound cells did not change, 8 mg/L; for P. aeruginosa FADDI-PA006 (polymyxin B MIC 8 mg/L), the polymyxin MIC of the rebound cells increased to 32 mg/L. These results suggest that the rebound growth seen at 24 hours with P. aeruginosa FADDI-PA006 is likely because of polymyxin B resistance, whereas with K. pneumoniae FADDI-KP001 and A. baumannii FADDI-AB156, the regrowth at 24 hours is likely because of resistance to the SERM.
Antipseudomonal activity of the polymyxin B–toremifene combination in sputum
The antipseudomonal activity of polymyxin B and toremifene per se and in combination against the P. aeruginosa isolate FADDI-PA067 (polymyxin B MIC 32 mg/L; toremifene >128 mg/L) was assessed in an ASM assay (Fig. 3). Although each drug alone was completely inactive, the polymyxin B–toremifene combination remarkably reduced cell viability to ∼15% of the untreated control cells under sputum-like conditions.

Antimicrobial activity of the polymyxin B–toremifene combination against P. aeruginosa FADDI-PA067 in artificial sputum media. Data points are plotted as the mean ± SD of four independent measurements. The top right-hand corner inset graphics in each bar graph show the raw data from the resazurin fluorescence plate assay; numbers 1–4 correspond to the numbering for each condition in the adjacent bar graph. The replicates for each condition are organized vertically in the 96-well plate.
Imaging the OM permeabilizing activity of polymyxin B–toremifene combination against P. aeruginosa
SEM and TEM imaging of P. aeruginosa FADDI-PA067 cells treated with polymyxin B and toremifene alone and in combination for 2 hours demonstrated extensive ultrastructural changes (Fig. 4). Our TEM images revealed that treatment with each compound alone or in combination produces ultrastructural damage to P. aeruginosa cells, including severe damage of the cell envelope, leakage of cytoplasmic material, and coagulation of the cytoplasmic matrix. In the SEM images, the treated cells appeared swollen and corrugated with membrane blebbing. These structural alterations clearly indicate a loss of cellular integrity. Similarly, confocal imaging of the P. aeruginosa strain AH298-GFP proved that the combination was more effective at producing damage to the cell envelope than each drug alone, as measured by the release of cytosolic GFP (Fig. 5).

Electron microscopy images of the P. aeruginosa isolate FADDI-PA067 (polymyxin B MIC 32 mg/L; toremifene >128 mg/L) treated with the polymyxin B–toremifene combination.

Top panels: confocal microscopy images of cytosolic GFP release from P. aeruginosa AH298-GFP treated with the polymyxin B–toremifene combination. Bottom panel: Bar graph showing the GFP signal as the percentage of the untreated control (n = 4). GFP, green fluorescence protein.
The polymyxin B–toremifene combination induces increased nitrocefin uptake across the OM and production of ROS in P. aeruginosa
To further confirm that the combination of polymyxin B–toremifene induced cell envelope damage, the nitrocefin assay was employed using the β-lactamase positive strain P. aeruginosa FADDI-PA067 (Fig. 6A). The assay measures periplasmic β-lactamase activity using nitrocefin, a chromogenic cephalosporin substrate analogue, which ordinarily permeates the OM slowly. 34 In the presence of OM permeabilizing agents, nitrocefin can more readily penetrate the OM and is hydrolyzed to its colormetric form by intracellular β-lactamase. The combination of polymyxin B–toremifene was significantly more effective at permeabilizing the OM than either of the drugs per se, seen from the increased β-lactamase-mediated conversion of nitrocefin (Fig. 6A).

ROS production is believed to be a common mechanism, whereby bacteriacidal antibiotics induce bacterial killing.35–37 Figure 6B shows that the combination induced a greater increase of ROS production in P. aeruginosa FADDI-PA067 cells than the treatment with each drug alone, as seen by the increase in fluorescence emission of the oxidative stress-sensitive dye CellROX Green.
The polymyxin B–toremifene combination induced cytoplasmic membrane depolarization in P. aeruginosa
The combination of toremifene and polymyxin B (16 μg/ml of each compound) showed a stronger membrane depolarizing effect on P. aeruginosa FADDI-PA067 cells than treatment with each compound alone (at 16 μg/ml) (Fig. 6C). Notably, toremifene displayed a significantly greater membrane depolarizing effect than polymyxin B. Triton 10 × at 0.5% was used as a positive control and carbapenem, meropenem, as a negative control of membrane depolarization. Meropenem shows no significant fluorescent changes as expected with the mode of action targeting the inhibition of cell wall synthesis, rather than being membrane active.
Discussion
The dawn of the “postantibiotic era” is approaching because of the rise of antibiotic-resistant “superbugs” and the fact that the pharmaceutical industry has not developed new antibiotic classes for more than a decade. Resistance to the last-line polymyxins is of particular concern, as bacteria can now rapidly acquire polymyxin resistance through the wide-spread dissemination of the mcr-1 plasmid.38–40 Repurposing of compounds from the vast FDA-approved drug reservoir is a highly viable, low-risk strategy for the development of novel antimicrobial therapies, particularly compared with de novo drug discovery. 4
This study demonstrates that synergistic antibacterial activity against problematic XDR Gram-negative “superbugs” can be found when combining antineoplastic SERM drugs with polymyxin B. Notably, the polymyxin B–SERM combinations achieved 65% synergy coverage across the 75 isolates tested, which is most encouraging. The less than complete coverage is likely because of variability in susceptibility to the antibacterial mechanisms involved across Gram-negative isolates. The SERM–polymyxin combinations produced excellent antibacterial killing kinetics against polymyxin-resistant P. aeruginosa, A. baumannii, and K. pneumoniae and very potent antipseudomonal activity under sputum-like conditions (Figs. 2 and 3).
SERMs are nonsteroidal estrogen receptor antagonists mostly used for chemotherapy and chemoprevention of breast cancers.11–13 Besides their antineoplastic activity, SERMs have been found to possess direct antimicrobial activities.15–18 Tamoxifen and toremifene have been shown to possess potent activity against Cryptococcus neoformans and Candida albicans by interfering with calcineurin signaling through binding to calmodulin and calmodulin-like proteins.15,17 With respect to the antifungal mode of action of SERMs, multiple mechanisms have been inferred, including membrane damage, inhibition of membrane peroxidation, cell cycle arrest, and, as already mentioned, interference with calcium homeostasis.15,17 Moreover, toremifene has been reported to display direct inhibitory activity against Ebola virus and inhibits biofilm formation by Candida spp., P. aeruginosa, Staphylococcus aureus, and Staphylococcus epidermidis.18,41–43 Raloxifene has been shown to diminish P. aeruginosa virulence in a Caenorhabditis elegans infection model. 41 Furthermore, raloxifene treatment produces a dose-dependent reduction in pyocyanin production by P. aeruginosa PAO1 and PA14. 41
The Gram-negative OM is a formidable barrier, especially against hydrophobic drugs such as the SERMs.
44
The antimicrobial action of polymyxins is mediated through a direct interaction with the lipid A component of the lipopolysaccharide (LPS), which leads to a disruption of the OM barrier.
6
The cationic
Notably, the nonapeptide–tamoxifen combination was inactive against the two polymxyin-resistant P. aeruginosa strains tested; this is likely because of the requirement for added hydrophobicity within the polymyxin scaffold to permeabilize the OM of resistant isolates. 46 However, it is unlikely that a single mode of action is responsible for the antibacterial synergy, particularly in view of the aforementioned multitude of cellular effects that SERMs mediate. An alternative mechanism, which we purport, is that the antibacterial synergy of the combination is also a result of their combined OM activity. As already mentioned, the primary mode of action of polymyxins involves disruption of the Gram-negative OM, similarly the SERMs are considered to be membrane-active drugs.47–50 Tamoxifen is known to interact with lipids in biomembranes and cause ultrastructural changes, which may result in cell lysis.43,48 Notably, triaryl butane analogues of tamoxifen have been shown to induce sodium and potassium efflux from Gram-negative cells and loss of transmembrane potential. 47
Similarly, our findings demonstrated that the polymyxin B–toremifene combination and toremifene treatment per se produce an appreciable depolarization of the cytoplasmic membrane of P. aeruginosa cells (Fig. 6C). Additional effects of tamoxifen that have been suggested to reflect its direct action on biomembranes include its ability to change the morphology of the breast tumor cell membranes 48 ; other biomembrane-related effects include hemolysis, 49 mitochondrial swelling, 51 and proton leakage from the mitochondrial inner membrane, resulting in depolarization of membrane potential.52,53 Accordingly, the accentuated ultrastructural alterations we observed in P. aeruginosa cells treated with the polymyxin B–toremifene combination compared with treatment with either of the drugs alone are potentially a consequence of their combined disruptive membrane activity (Fig. 4). Similar to our SEM and TEM findings, electron microscopy studies of the membrane effects of tamoxifen on a strain of Bacillus stearothermophilus revealed changes in the membrane structure from asymmetric to symmetric and the appearance of fractures in the cell wall. 50 The synergistic damaging effect of the polymyxin B–toremifene combination was corroborated by the GFP release and nitrocefin assay data (Figs. 5 and 6A). Notably, both polymyxin B and toremifene alone and in combination produced a marked increase in cellular ROS in P. aeruginosa cells, which suggests that the latter could also contribute to the potent killing effect (Fig. 6B).
In summary, our study demonstrates that the combination of polymyxin B with SERMs might offer a new opportunity to treat otherwise untreatable XDR Gram-negative infections. P. aeruginosa in particular is intrinsically resistant to antibiotics, and is noted as requiring an urgent need for new antipseudomonal therapeutic options. 54 From a practical standpoint, this in vitro study suggests that the “off-the-shelf” SERMs represent a potentially novel antibiotic class for further antibiotic drug discovery and development against multi-drug resistant (MDR) Gram-negative “superbugs.” The SERM concentrations used in this study are achievable in patients through oral administration and are well tolerated.55–58 The high oral bioavailability of tamoxifen and toremifene together with the availability of pre-existing libraries of analogues from structure–activity relationship studies performed during their development as breast cancer therapeutics could expedite the identification of novel SERMs with superior antibacterial activities.
Footnotes
Acknowledgments
J.L. and T.V. are supported by the Australian National Health and Medical Research Council (NHMRC). We want to thank Maite Amado for her technical assistance in running the DisC3-5 assay.
Disclosure Statement
No competing financial interests exist.