
Abstract
The Enterobacteriaceae is a large family of Gram-negative, facultative anaerobic, non-spore forming rod-shaped bacteria that includes harmless and pathogenic organisms. The emergence and development of drug resistance in Enterobacteriaceae is complicating the treatment of serious infections. The aim of this study is to predict and characterize putative drug targets in Enterobacteriaceae family employing a homology-based computational method. The final putative drug targets were qualitatively characterized via cellular function prediction, subcellular localization prediction, broad-spectrum, and druggability analyses. Of 6,327 analyzed proteins, 35 proteins were selected as final putative drug targets in Enterobacteriaceae family. These putative drug targets were involved in different vital pathways like metabolism, biosynthesis of macromolecule, and cell division. Predicted drug targets were also localized in the cytoplasm and cytoplasmic membrane of the pathogen that acts as antimicrobial or vaccine targets. Of 35 drug targets, 5 targets were druggable and 30 targets were not druggable and were predicted as novel drug targets, which should be further evaluated to develop new antimicrobial. Thirteen drug targets were considered as broad-spectrum targets. It is expected that results of our study could facilitate the production of novel antibacterial for efficient treatment of infections caused by Enterobacteriaceae pathogens.
Introduction
M
In contrast, in silico identification methods could speed up the drug discovery procedure, increase therapeutic targets, provide a closer look of the entire microorganism at a time and reduce drug defeat rate in the clinical trials. Therefore, the application of in silico identification methods for finding drug targets has been on the increase in drug discovery investigations.3–5 Several in silico methods can be employed for discovery of a proper therapeutic target like models based on comparative genomics, 6 structure and sequence to function,7,8 metabolic pathways, 9 and data mining. 10
Subtractive and comparative genomic methods based on the sequential alignment against host proteome and essential genes database, have been used to predict drug targets in bacterial pathogens like Leptospira interrogans, 11 Helicobacter pylori, 12 Mycobacterium tuberculosis and M. ulcerans,4,5,13 Burkholderia pseudomallei, 14 Aeromonas hydrophila, 15 and Pseudomonas aeruginosa.16,17 Ligand-docking and structure-based methods have been applied to identify potential drug targets in influenza A virus, 7 Plasmodium falciparum, 8 and M. tuberculosis. 18
Structure-based methods use the three-dimensional structure, ligand-docking, ligand and receptor interaction information of proteins to predict specific targets. The methods that compare metabolic pathways have been effectively applied to identify potential drug targets in P. falciparum,9,19 M. tuberculosis,19,20 and Staphylococcus aureus. 21 In the drug target discovery process, data mining approaches combine biological concepts with computer tools or statistical methods to discover potential drug targets.22,23 Data mining methods have successfully discovered potential drug targets in Escherichia coli, 10 M. tuberculosis,24,25 and Aspergillus fumigatus. 26
Most of these drug target discovery approaches consider essentiality or specificity as the main properties for potential drug candidates. Beside these criteria, a suitable drug target must be specific to the pathogen for the avoidance of harmful side effect and should be a vital protein for survival of the pathogen. Inhibition of such drug targets can result in effective control of the pathogen without any harmful effect on the host.4,5,13
Three other important parameters that should be evaluated for a suitable drug target are druggability, broad-spectrum condition, and subcellular localization. The site of accumulation of target proteins in the cell is necessary to determine the type of targeting strategy. Proteins located on extracellular surfaces (membrane and extracellular proteins) and intracellular proteins (cytoplasmic proteins) can be used as vaccine and drug targets, respectively. Broad-spectrum condition of a drug target protein can be used to develop broad-spectrum or specific antimicrobials. Druggability is one of the most important parameters of a target molecule. A druggable target binds with high affinity to drug molecules and such target can be quickly used in drug discovery process, while novel drug targets should be further evaluated before entering the drug discovery procedures.
In this study, we use a homology-based method for genome-wide prediction of the drug targets in the major pathogens of Enterobacteriaceae family considering the Shigella flexneri (S. flexneri) as an important pathogen of this family (Fig. 1).

The complete flowchart of the homology-based method.
Materials and Methods
First stage
The complete proteome of S. flexneri 2a str. 301 comprises 4,053 chromosomal proteins and 262 plasmid proteins were downloaded from NCBI genome database. Virulence factor database (VFDB) is a practical database of Virulence factors (VFs) for bacterial pathogens and also Chlamydia and Mycoplasma. All the major VFs and pathogenicity islands (PAIs) proteins of S. flexneri (188 proteins) were retrieved from VFDB. All literature reported S. flexneri resistance proteins were downloaded from NCBI.1,5
Using STRING 10.0 tool and seven active prediction methods (neighborhood, gene fusion, co-occurrence, co-expression, experiments, databases, and text-mining), interaction partners of S. flexneri resistance proteins with high confidence value (0.7) were predicted. The protein sequences of resistance genes and their partners were retrieved from NCBI and STRING database. Finally, a comparative metabolic pathway analysis between S. flexneri and human as the host was carried out.
All the proteins in distinct pathways of S. flexneri and unique proteins of S. flexneri in common pathways (between S. flexneri and human) were collected. All proteins in distinct pathways of human and unique proteins of human in common pathways were excluded. The amino acid sequences of the selected proteins were downloaded from KEGG and NCBI. Total proteins of the first stage (greenlist) were transferred to the second stage.
Second stage
Host homology search
The goal of this search is to find S. flexneri specific proteins that are not homologous with human proteome. This analysis minimizes the undesirable side effects of drugs.5,27 The greenlist proteins were subjected to BLASTp against non-redundant protein sequences of human (taxid: 9606) with an e-value of 0.005.5,27,28 The greenlist proteins that showed no hits for the 0.005 e-value were transferred to next test (test of essentiality).
Test of essentiality
The proteins in greenlist were examined to recognize essential proteins using BLASTp search against Database of Essential Genes (DEG). The DEG BLASTp expected value cutoff was adjusted to 0.00001. The hits with less than e-value of 0.00001, identity ≥25%, and same annotated function of the query were selected as essential proteins.5,13,27 The greenlist protein that exhibited hits with DEG BLASTp parameters were collected and transferred to next step (gastrointestinal flora homology test). Other proteins that exhibited no hit in DEG BLASTp were excluded from the analysis.
Gastrointestinal flora homology search
Unintentional targeting of vital molecules in the gut microorganisms may cause harmful effects on host. To prevent this condition, a custom database (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/mdr) was created from gut microflora reported in literature and the proteins of greenlist were subjected to BLASTp search against this custom database with an expected threshold of 0.0001.4,5 The greenlist proteins that demonstrated more than 10 hits were excluded and the rest were selected as whitelist. The whitelist comprises the primary potential drug targets of S. flexneri.
Third stage
Whole proteome comparisons
To find similarities, proteome of nine important pathogens of Enterobacteriaceae (Escherichia, Shigella, Salmonella, Klebsiella, Enterobacter, Serratia, Citrobacter, Proteus, and Yersinia) were compared. All genomes belonging to these eight genera were compared in TaxPlot with a cutoff value of 200.
Enterobacteriaceae homology search
To find homologs in important pathogens of Enterobacteriaceae, primary potential drug targets of S. flexneri in whitelist, were subjected to BLASTp against custom database (Supplementary Table S2) of nine genera (Escherichia, Shigella, Salmonella, Klebsiella, Enterobacter, Serratia, Citrobacter, Proteus, and Yersinia). The BLASTp was performed with an expected value of 0.0001. Proteins with identity ≥50% were selected as final potential drug targets in Enterobacteriaceae family (redlist) and were qualitatively characterized using various tools in the fourth stage.
Fourth stage
The biological function of unknown protein in redlist (hypothetical proteins) was determined by means of Pfam. Subcellular localization sites of some proteins were recognized in literature, and web-based tools, such as PSLpred, PSORT, and CELLO, were used to predict possible subcellular localization sites of redlist proteins. In the Broad-spectrum search, a custom database (Supplementary Table S3) of bacterial pathogens reported in literature 4 was created. Proteins in redlist were evaluated by BLASTp search against custom database with an e-value of 0.005 for the identification of broad-spectrum targets. In DrugBank database, 29 redlist proteins were subjected to BLASTp search with an e-value of 0.00001. The presence of molecule with same biological function in BLAST results indicates druggability of the target molecules while absence shows the novelty of the target molecules.5,13
Results
First stage
In the complete proteome (chromosomal and plasmid) of S. flexneri, 2,175 proteins are produced by negative-strand and 2,140 proteins are translated from positive-strand. Distributions of the protein lengths in the S. flexneri proteome were in the range of 21–1,522 amino acids (Fig. 2A). The proteins of S. flexneri were clustered into 12 functional categories. Proteins with catalytic activity were dominant category alongside proteins with binding and transporter activity (Fig. 2B). Major VFs in S. flexneri have been divided into the following six groups: actin-based motility system, endotoxins, iron uptake system, proteases, secretion systems, and toxins.
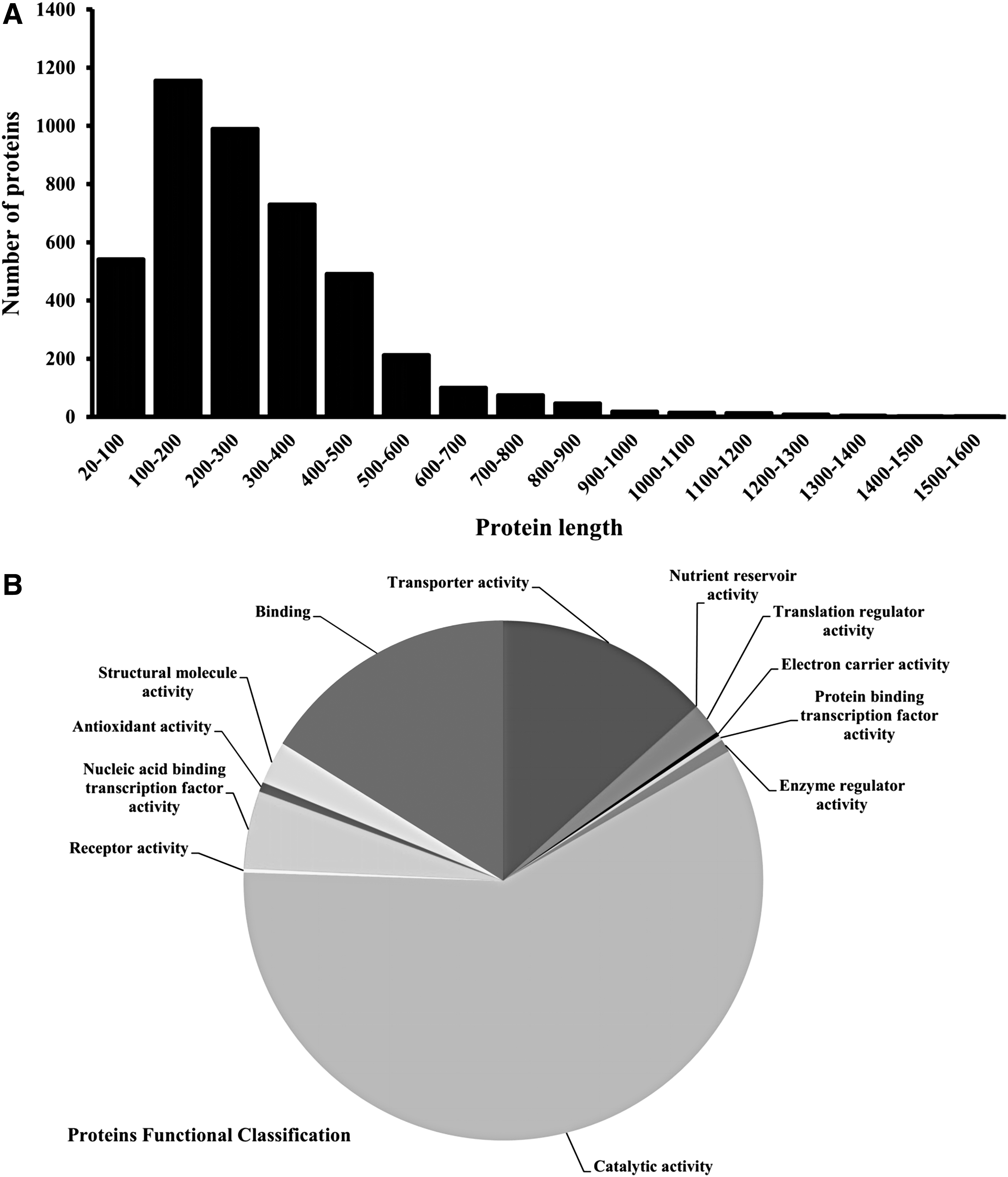
Complete proteome analysis of Shigella flexneri.
Also, four PAIs have been identified in S. flexneri: SHI-1, SHI-2, SHI-3, and SRL. PAIs and VFs have been located on S. flexneri chromosome and pCP301 plasmid. The SHI-1, SHI-2, SHI-3, and SRL PAIs size were 46, 24, 21, and 66 kb, respectively and GC content of these PAIs were largely similar to host (S. flexneri). Eighteen proteins that confer resistance to antimicrobial drugs in S. flexneri were obtained from literatures and corresponding sequences retrieved from NCBI. Using STRING 10.0 tool, 469 interaction partners for resistance causing proteins were predicted. In KEGG database, there are 299 metabolic pathways for human and 115 metabolic pathways for S. flexneri. Of 115 metabolic pathways in S. flexneri, 35 pathways were unique and 80 pathways were common with human. Sequences of 945 unique proteins from common pathways and 392 proteins from distinct pathways were selected.
Finally, 6,327 proteins including chromosomal and plasmid proteome, VFs, PAIs, resistance proteins, and their interaction partners, common and unique metabolic pathways proteins were collected as greenlist and transferred to second stage.
Second stage
A collection of 6,327 proteins that resulted from the first stage (greenlist) were subjected to three sequential BLASTp searches to finding primary targets (Fig. 1). Of 6,327 input proteins, 1,495 proteins that demonstrated no hits against human proteome were selected and 4,832 proteins (homologous to human proteome) were excluded from the next stage of analysis. The greenlist proteins were analyzed for the determination of essentiality using DEG 11.3 database. Of 1,495 input proteins, 874 proteins were identified as essential for the pathogen and selected for the next analyses. In addition, 621 proteins that revealed no hits against DEG (non-essential) were excluded from the analysis. The resulting essential proteins from previous analyses were subjected to BLASTp against custom database of the whole proteome of gut microbiota. Of 874 input proteins, 125 proteins were collected as whitelist and 749 proteins exhibiting more than 10 hits were excluded. Whitelist proteins were selected as primary potential drug target of S. flexneri and used for next stage of analysis.
Third stage
The reference proteome of S. flexneri was compared with Enterobacteriaceae pathogens to find similarities. Whole proteome comparisons of nine important pathogens of Enterobacteriaceae demonstrated high similarity among this family. In TaxPlot analysis, S. flexneri proteome exhibited 88% similarity to all the species of Escherichia genus, 96% to Shigella genus, 75% to Salmonella genus, 74% to Klebsiella genus, 79% to Enterobacter genus, 71% to Serratia genus, 77% to Citrobacter genus, 60% to Proteus genus, and 69% to Yersinia genus. In the Enterobacteriaceae homology search, the whitelist were subjected to BLASTp against custom database of nine important genera in Enterobacteriaceae family, to find homologs. From 125 proteins in the whitelist, 35 proteins that showed identity ≥50% were selected as redlist (Table 1). Redlist proteins were regarded as final potential drug targets in Enterobacteriaceae family (Table 2).
Fourth stage
In this stage, redlist proteins were further explored using various analyses (functional search, subcellular localization search, broad-spectrum search, and druggability search) (Fig. 1).
Eight proteins in redlist, namely, AAL08478.1, SF1007, SF2371, SF2999, SF4226, SF0611, SF1770, and SF1857 were unknown and hypothetical (Table 2). Unknown protein AAL08478.1 and hypothetical protein SF2999 were determined to be CbtA toxin, which is described to be involved in type IV toxin–antitoxin system. Hypothetical protein SF1770 was predicted to be an YdcZ inner membrane exporter. YdcZ is a family of putative exporter proteins from both Gram-negative and Gram-positive bacteria. Hypothetical protein SF0611 was determined to be an YbfN-like lipoprotein. This family of proteins is found in bacteria and is functionally uncharacterized. Hypothetical protein SF1007 was predicted to be an YccJ-like protein that is functionally uncharacterized. The YccJ-like family of proteins is found in bacteria.
Members of this family are ∼80 amino acids in length. Hypothetical proteins SF2371, SF1857, and SF4226 were domains of unknown function, which are protein domains that have no characterized function. In PSORTb and PSLpred, based on the localization score, reliability index, and expected accuracy, 16 targets were predicted to be cytoplasmic proteins, 9 targets were predicted to be cytoplasmic membrane proteins and 1 target was predicted to be outer membrane protein. The cellular localization of nine targets (SF1007, SF2999, SF3844, SF4226, SF4381, SF4353b, SF0611, SF1944, and SF3362) was predicted to be unknown. In CELLO, the location of 22 targets was determined as cytoplasmic, 12 as inner membrane, and 1 as extracellular. Of nine targets that are characterized as unknown by PSORTb and PSLpred, six targets were predicted as cytoplasmic and three targets as inner membrane (Table 3). Results of broad-spectrum search indicated that 13, 10, and 12 targets were present in more than 100, between 50 and 100, and <50 pathogens, respectively. Thirteen proteins of redlist that were present in more than 100 pathogens were considered as broad-spectrum targets (Table 3). Four targets that are present in <25 pathogens were considered as Enterobacteriaceae specific targets. Similarity search revealed that five targets (SF0250, SF0575, SF1479, SF4020, and SF4309) are homologous to one or more known targets in DrugBank database with an expected value of 0.00001.
DUF, domains of unknown function.
BLASTp against DrugBank also revealed that SF0250 target has one experimental inhibitor (2-aminoethanesulfonic acid and (2R)-2-ethyl-1-hexanesulfonic acid), SF0575 has three experimental inhibitors (atpenin A5, 2-[1-methylhexyl]-4, 6-dinitrophenol, and ubiquinone-2), SF1479 has one experimental inhibitor (formic acid), SF4020 has one experimental and two approved inhibitors (2-amino-3-(1-hydroperoxy-1h-indol-3-yl)propan-1-ol, ethionamide and isoniazid), and SF4309 has two experimental inhibitors (2-[1-(4-chloro-phenyl)-ethyl]-4,6-dinitro-phenol and 2-heptyl-4-hydroxy quinoline n-oxide). Thirty targets demonstrating non homologous with DrugBank database was considered as novel drug targets, which should be further evaluated experimentally (Table 3).
Discussion
Due to high adverse effects related to antibiotic treatments, development of an efficient target prediction method is necessary. In this study, a subtractive homology-based method was used to identify a collection of potential therapeutic targets in some bacterial pathogens of Enterobacteriaceae family. Only those proteins that successfully passed the pipeline were selected as acceptable drug targets (Fig. 1).
Among 35 final targets, AAL08478.1 and SF2999 are VF proteins that are involved in type IV toxin–antitoxin system. Toxin–antitoxin systems are involved in stress response, persistence, biofilm formation, and pathogenicity of bacteria. Suppression of toxin–antitoxin system proteins may lead to breaking the balance of toxin–antitoxin levels, disruption in physiological processes, loss of virulence, and induction of apoptosis in pathogen. 30
Most of the current antimicrobial drugs mainly target proteins from DNA, protein, and cell wall biosynthesis machineries. 31 Currently, drug target discovery methods are focused on other vital pathways like lipopolysaccharide and peptidoglycan biosynthesis pathways. Restricted targeting of some specific pathways may cause development of multidrug resistance among pathogenic bacteria. 5 Generally, approaches that consider all vital pathways of organisms can be more successful for the identification of efficient drug targets. Proteins involved in several pathways are efficient drug targets because inhibition of such proteins leads to disruption of many biological processes. In addition, targeting of cell division proteins and regulatory proteins could reduce virulence, growth, and development of microorganisms.
The redlist consists of proteins involved in metabolism, biosynthesis of macromolecule, oxidative phosphorylation, citrate cycle, two-component system, quorum sensing, protein export, bacterial secretion system, glycolysis, gluconeogenesis, pentose phosphate pathway, glycan degradation, sulfur relay system, cellular transport, cell division, degradation of xenobiotics, regulatory processes, signal transduction, mismatch repair, homologous recombination, and other metabolic pathways.
By means of two-component system, the pathogens perceive the changes in the environment and respond to it. Also, involvement in pathogenicity of the organisms and the absence of these systems in human makes them attractive drug targets. 32 The two targets (SF1229 and SF4309) are found in two-component system. Therefore, inhibition of these proteins could decrease growth and virulence of the pathogen.
Targeting of proteins involved in DNA replication, DNA repair, DNA recombination, and cell division (SF0606, SF0919, SF0920, SF2070, SF2833, and SF4353b) may disrupt the pathways necessary for pathogen survival, growth, and reproduction.
Of 35 redlist targets, 15 targets (SF0250, SF0575, SF1229, SF1479, SF3648, SF3817, SF3844, SF4003, SF4020, SF4381, SF0370, SF1944, SF3117, SF3915, and SF4309) were involved in more than one metabolic pathway (Table 2). Targets involved in multiple metabolic pathways are thought to be more efficient drug targets and preventing the activity of such targets could increase lethal effects by blocking the activity of several metabolic pathways of the microorganism.
Thirteen broad-spectrum targets are involved in crucial process such as metabolism, two-component system, DNA replication, DNA recombination, and DNA repair (Tables 2 and 3). Drug targets involved in vital metabolic process appear to be broader spectrum than the other targets such as VFs. Due to high specificity, VFs are not often broad-spectrum drug targets. Targeting of broad-spectrum proteins by drug molecules may facilitate the destruction of wide range of pathogenic bacteria. Four Enterobacteriaceae specific targets are suitable for development of narrow spectrum antibiotic. Such specific target proteins may reduce the threat of development of antimicrobial resistance in wide range of pathogenic bacteria.
Proteins located on membrane and extracellular proteins can be used as vaccine targets, whereas cytoplasmic proteins can act as drug targets. Twenty-two proteins with cytoplasmic localization and 13 proteins with inner membrane and extracellular localization could possibly serve as drug and vaccine targets, respectively (Table 3). Inhibition of target proteins located on membrane and extracellular proteins is important due to their crucial role as VFs aiding pathogens to spread and proliferate within the host. Despite the trouble of a drug molecule crossing the cytoplasmic membrane, our study demonstrated a significant preference for intracellular drug targets, perhaps because proteins are abundant in the cytoplasm.
Druggability refers to the ability of a target molecule to bind with high affinity to the drug molecules. Druggability is one of the most important characteristics of a target molecule. Five druggable targets (SF0250, SF0575, SF1479, SF4020, and SF4309) are involved in vital process like metabolism and biosynthesis of macromolecule. Either of these proteins is a target for conventional antibiotics. Thirty novel drug targets are suitable for development of new antimicrobials and should be further evaluated experimentally.
Currently, several computational methods such as comparative genomics, data mining, structure and sequence to function, and metabolic pathways are utilized for identification of potential drug targets. These approaches consider specificity or essentiality as the main criteria for potential drug candidates. Beside these properties, a suitable drug target must be specific to the pathogen to avoid harmful side effects and should be a vital protein for survival of the pathogen. Suppression of such drug targets can result in effective control of the pathogen without any harmful effects on the host. Our homology-based method considers the essentiality, specificity, druggability, subcellular localization, and function and broad-spectrum condition of drug targets. Although sequence similarity of protein does not ensure the same structures or binding properties, using such homology-based methods could ease the optimization and production of new drugs and vaccines.
Footnotes
Acknowledgments
The authors would like to acknowledge the Bioinformatics Lab, School of Medicine, and Kerman University of Medical Sciences for their support during the course of this work.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.