
Abstract
Imipenem with relebactam is a novel β-lactam-β-lactamase inhibitor that has activity against most KPC-producing Enterobacteriaceae. Using 10 isolates of KPC-possessing Klebsiella pneumoniae, we assessed the relationship between imipenem–relebactam minimum inhibitory concentrations (MICs) and mechanisms known to contribute to antimicrobial resistance. The effect of adding a second agent was assessed by time-kill experiments. Mutations affecting the genes encoding porins ompK35 and ompK36 and identification of β-lactamases were assessed by PCR. Expression of blaKPC and acrB was assessed by real-time reverse-transcriptase (RT)-PCR, and production of OmpK36 by SDS-PAGE. Time-kill studies were performed using combinations of imipenem–relebactam with polymyxin B, amikacin, or tigecycline. Seven isolates having a disruption in a single porin, or in neither porin, remained susceptible to imipenem–relebactam. The addition of a second agent did not improve the activity of imipenem–relebactam for these isolates, although the addition of tigecycline was antagonistic for three isolates. Three isolates had major disruptions in both ompK35 and ompK36 that correlated with reduced activity of imipenem–relebactam (MICs 2/4, 8/4, and 512/4 μg/mL). Two of these isolates also had overexpression of blaKPC, including the isolate with the highest MIC. These isolates were also resistant to polymyxin B and amikacin. The addition of amikacin provided both synergistic and bactericidal activity for the two more resistant isolates. The activity of imipenem–relebactam against K. pneumoniae is affected by major disruptions of both ompK35 and ompK36 and by expression of the KPC gene. Combining imipenem–relebactam with an aminoglycoside may be a promising approach for isolates with reduced susceptibility to imipenem–relebactam.
Introduction
M
Relebactam, avibactam, and vaborbactam are among several novel β-lactamase inhibitors developed with enhanced activity against class A and class C enzymes. Previous studies have demonstrated that in the presence of 4 μg/mL of relebactam, nearly all isolates of KPC-positive Enterobacteriaceae are inhibited by 1 μg/mL of imipenem.4,5 Resistance of KPC-possessing isolates to these newer β-lactamase inhibitors has been reported, although the mechanisms contributing to resistance have not been fully determined.6–10 In one report involving KPC-possessing K. pneumoniae, the combined presence of porin mutations and an ESBL was associated with higher ceftazidime–avibactam minimum inhibitory concentrations (MICs) (in the 1–4/4 μg/mL range). 9 In this study, we examine the effect of mutations involving porins ompK35 and ompK36 and expression of blaKPC on the activity of imipenem–relebactam and whether antibiotic combinations can overcome any reduction in activity.
Materials and Methods
Bacterial isolates
Ten clinical isolates of KPC-positive K. pneumoniae were selected from surveillance studies conducted at Brooklyn, NY hospitals between 2004 and 2014. MICs were performed in triplicate by the broth microdilution method; for imipenem–relebactam, relebactam concentrations were constant at 4 μg/mL. Multilocus sequence typing was performed according to established methods. 11 Isolates were selected to represent several unique strains as well as several isolates from the dominant sequence type in the region with a range of susceptibility to imipenem–relebactam.
Time-kill studies
Time-kill experiments were performed in cation-supplemented Mueller-Hinton broth as previously described with a starting inoculum of ∼1 × 106 cfu/mL. 12 Antibiotic concentrations tested were imipenem–relebactam 4/4 μg/mL, polymyxin B 2 μg/mL, amikacin 16 μg/mL, and tigecycline 1 μg/mL. Antibiotic carryover was eliminated by a 400-fold dilution in pour plates. Synergy and antagonism were defined as a ≥100-fold increase or decrease, respectively, in killing at 24 hours by an antibiotic combination compared with each individual agent. Bactericidal activity was defined as a ≥3 log cfu/mL decrease in colony count at 24 hours.
Mechanisms of resistance
Previously described primers and conditions were used to identify SHV and TEM β-lactamases as well as ompK35 and ompK36. 13 The presence of carbapenemases (VIM, IMP, NDM, OXA, and KPC) was determined using previously described primers and conditions (5). Outer membrane proteins were examined by SDS-PAGE; protein migration patterns were previously identified by matrix-assisted laser desorption/ionization mass spectrometry. 13 The expression of blaKPC and the efflux gene acrB were determined by real-time reverse-transcriptase (RT)-PCR as previously described. 13 RT-PCR experiments were performed in triplicate using primer and probe concentrations that resulted in amplification efficiencies of 90–110%. The calibrator for blaKPC expression was the isolate with the lowest level of expression and for acrB expression was K. pneumoniae ATCC 11296. Most isolates had acrB expression that was 1–3 times that of the K. pneumoniae ATCC 11296; isolates with expression >4 times K. pneumoniae ATCC 11296 were considered to have overexpression. Most isolates had blaKPC expression 1–15 times that of the isolate with the lowest expression; overexpression was considered when there was ≥15 times that of the isolate with the lowest expression. Isolates were initially grouped and analyzed based on gene sequences and SDS-PAGE indicating disruption of neither, one, or both of the porin genes ompK35 and ompK36.
Results
Susceptibility and time-kill studies
Ten isolates of KPC-producing K. pneumoniae were selected with imipenem–relebactam MICs ranging from 0.25 to 512/4 μg/mL (Table 1). None of the isolates was found to harbor other carbapenemases. Most isolates belonged to ST258, while the remaining isolates were all unrelated (Table 1). As noted, isolates were initially grouped based on expression of the porins.
MIC, minimum inhibitory concentrations; ST, sequence type.
As a baseline comparator, one clinical isolate lacking blaKPC was chosen. This isolate (VA302) was multilocus sequence type 234 and possessed blaSHV-5 and blaTEM-1. There were no insertions, deletions, or frameshift mutations involving ompK35 and ompK36; ompK35 was wild type, and ompK36 possessed 12 amino acid substitutions. This isolate was quite susceptible to imipenem and imipenem–relebactam (MICs 0.12 and 0.12/4 μg/mL, respectively).
Group one isolates
Group one isolates had wild-type ompK35 (Table 1). Regarding OmpK36, all the isolates had several amino acid substitutions commonly seen in nosocomial isolates of K. pneumoniae from our area. These isolates also had short amino insertions involving OmpK36. Isolate KB41 possessed a six-amino acid insertion at amino acid 305. Isolate BD37 possessed a three-amino acid insertion at amino acid 183 and an Ala insertion at amino acid 229. Isolates KB528 and MA63 had a seven-amino acid insertion at amino acid 264. Isolate KB528 also possessed ins AA 135–136 GD that has been linked to carbapenem resistance. 14 All four had OmpK36 expressed by SDS-PAGE (Fig. 1). The four isolates had imipenem/relebactam MICs of 0.25/4 to 0.5/4 μg/mL and were susceptible to polymyxin B and amikacin. Expression of blaKPC was not increased for these four isolates. Similarly, acrB expression was not increased in the four isolates. The presence of an ESBL (SHV-12-like in isolate BD37) also did not appear to affect the MIC of imipenem–relebactam. In the time-kill experiments, bactericidal activity was achieved with imipenem/relebactam (4/4 μg/mL) for all four isolates, regardless of the addition of polymyxin B or amikacin. However, the addition of tigecycline was antagonistic for two of the isolates (KB528 and MA63).

SDS-PAGE of outer membrane proteins for the 10 isolates of Klebsiella pneumoniae.
Group two isolates
Three isolates had mutations leading to major disruption of ompK35 or ompK36. Two isolates (WO37 and DM44) possessed a frameshift mutation affecting OmpK35 at amino acid 42 (Table 1). Both isolates possessed similar OmpK36 phenotypes, with a three-amino acid insert at amino acid 183 and an Ala insertion at amino acid 229; OmpK36 was evident by SDS-PAGE (Fig. 1). The third isolate in this group, QU98, possessed a wild-type OmpK35 and had a premature stop codon within ompK36 at amino acid 144, and OmpK36 was not evident by SDS-PAGE (Fig. 1). Expression of acrB was increased in two isolates, but did not seem to affect imipenem–relebactam MICs. Similarly, the presence of an ESBL did not appear to affect the MICs. For these isolates, imipenem–relebactam concentrations of 4/4 μg/mL afforded bactericidal activity, with or without the addition of a second agent. However, antagonism was found with the addition of tigecycline for one isolate (WO37).
Group three isolates
Three isolates in this group had major disruption of both ompK35 and ompK36 and had imipenem–relebactam MICs of 2/4 to 512/4 μg/mL. All had a frameshift mutation affecting ompK35 at amino acid 42. Regarding OmpK36, none of the three isolates had production evident by SDS-PAGE (Fig. 1). Isolate IF19 had a transposase insertion at amino acid 14 and isolate ME9 had an IS4 sequence following amino acid 32 (Table 1). As with the other groups, expression of acrB did not appear to account for the variability in the MICs for imipenem–relebactam. Two isolates in this group (KC850 and ME9) had overexpression of blaKPC, including the isolate which was highly resistant to imipenem–relebactam (MIC 512 μg/mL). The three isolates were nonsusceptible to both polymyxin B (MIC ≥16 μg/mL) and amikacin (MIC 32 μg/mL). In the time-kill experiments, imipenem–relebactam (4/4 μg/mL) resulted in bactericidal activity only for isolate KC850 (MIC of imipenem–relebactam 2 μg/mL). For the other two isolates with higher imipenem–relebactam MICs, bactericidal activity was achieved only with the combination of imipenem–relebactam (4/4 μg/mL) and amikacin (16 μg/mL; Fig. 2A, B). The addition of tigecycline to imipenem–relebactam was not found to be antagonistic for any of the isolates in this group.
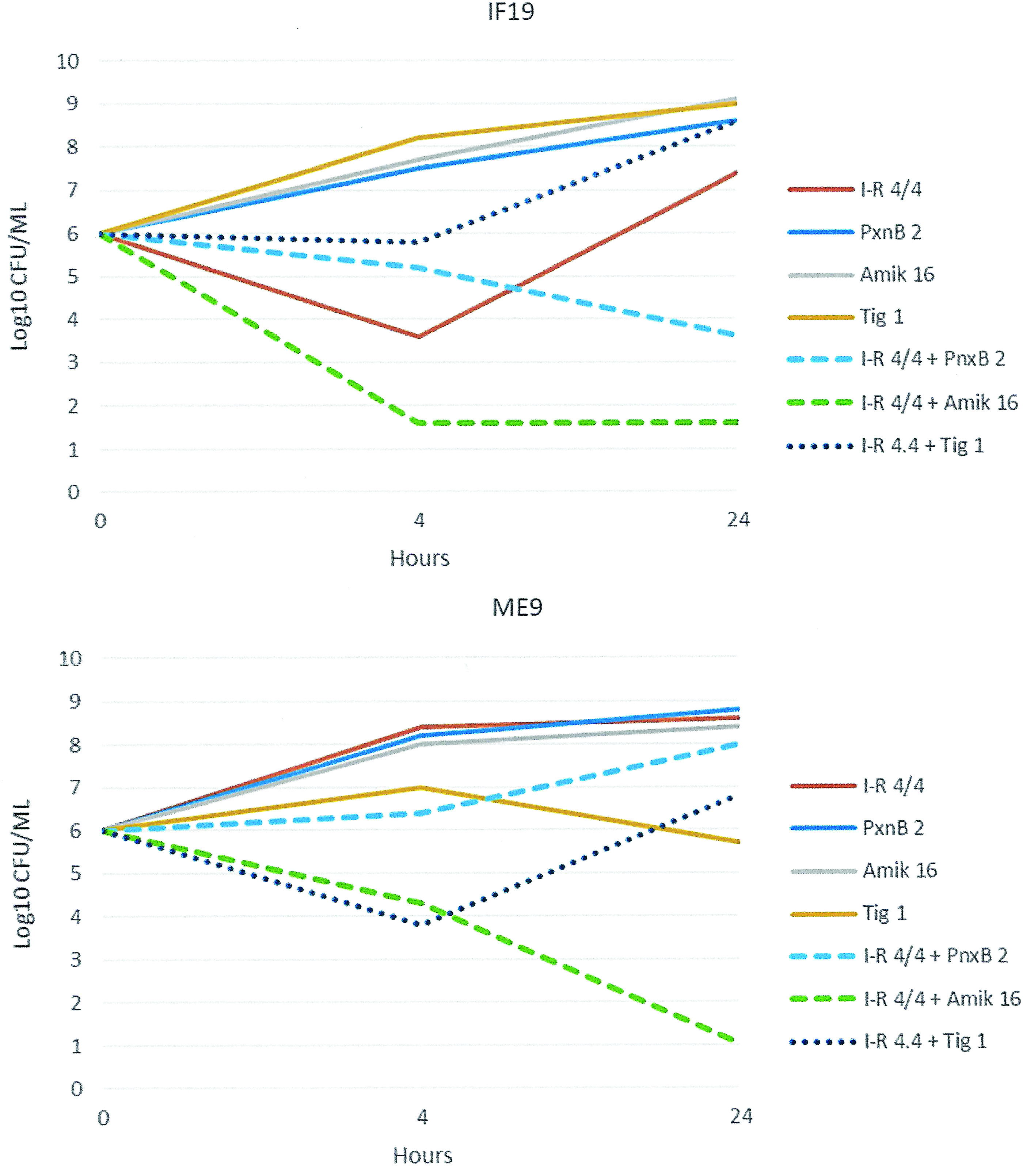
Time-kill experiments involving isolates IF19 and ME9, both with reduced susceptibility to imipenem–relebactam. I-R 4/4, Imipenem–relebactam 4/4 μg/mL; PxnB 2, Polymyxin B 2 μg/mL; Amik 16, Amikacin 16 μg/mL; Tig 1, Tigecycline 1 μg/mL. Color images available online at www.liebertpub.com/mdr
Discussion
The newer group of β-lactamase inhibitors, including relebactam, represents an important advance in our ability to manage infections due to KPC-producing pathogens. Reduced susceptibility to the novel β-lactamase inhibitor combinations has been rarely noted among KPC-producing Enterobacteriaceae. In a recent report, however, 3 of 37 patients treated for CRE infections with ceftazidime–avibactam developed resistance during therapy. 10 In these cases, the ceftazidime–avibactam MICs rose from 2–4 μg/mL to 128–>256 μg/mL. Information regarding factors that affect the activity of these agents is limited. Moreover, the optimal treatment strategies to maximize potential therapeutic efficacy and prevent the emergence of resistance are unknown.
In this group of KPC-producing K. pneumoniae, imipenem–relebactam MICs were not significantly affected by changes in expression of acrB, or the presence of SHV ESBLs. It also appears the disruption of either ompK35 or ompK36 alone did not affect MICs of imipenem–relebactam. However, when both were disrupted the MIC of imipenem–relebactam increased. The addition of high-level expression of blaKPC to dual porin alterations may also contribute to resistance to imipenem–relebactam. In a recent report, mutations involving ompK36 (but not ompK35) were linked to increased imipenem–relebactam MICs in KPC-possessing K. pneumoniae. 14 It is becoming increasingly evident that disruption of OmpK36 can contribute to resistance to imipenem–relebactam, and the role of OmpK35 requires further study. In our study, the finding of one isolate (ME9) with disproportionate resistance to imipenem–relebactam suggests that other factors are involved in resistance and warrants further study.
The time-kill studies demonstrated that imipenem–relebactam was bactericidal against the KPC-producing K. pneumoniae at the concentration of 4/4 μg/mL if the MIC was ≤4/4 μg/mL. This would include nearly all isolates from previous studies.4,5 The addition of tigecycline to imipenem–relebactam was antagonistic against some of these and pending further study should probably be avoided. For the two isolates with reduced susceptibility of imipenem–relebactam (8 and 512 μg/mL), the combination of imipenem–relebactam plus amikacin was synergistic and bactericidal. Limitations of our study include the concentrations chosen for the time-kill studies, which may be difficult to achieve clinically for most of the dosing interval. Nonetheless, the potential benefit of combining the imipenem–relebactam with aminoglycosides, in the face of reduced susceptibility to the former agent, deserves further study.