
Abstract
Background:
Klebsiella pneumoniae is a major opportunistic pathogen frequently associated with nosocomial infections, and often poses a major threat to immunocompromised patients. In our previous study, two K. pneumoniae (K36 and B13), which displayed resistance to almost all major antibiotics, including colistin, were isolated. Both isolates were not associated with infection and isolated from the stools of two preterm neonates admitted to the neonatal intensive care unit (NICU) during their first week of life.
Materials and Methods:
In this study, whole genome sequencing was performed on these two clinical multidrug resistant K. pneumoniae. We aimed to determine the genetic factors that underline the antibiotic-resistance phenotypes of these isolates.
Results:
The strains harbored blaSHV-27, blaSHV-71, and oqxAB genes conferring resistance to cephalosporins, carbapenems, and fluoroquinolones, respectively, but not harboring any known plasmid-borne colistin resistance determinants such as mcr-1. However, genome analysis discovered interruption of mgrB gene by insertion sequences gaining insight into the development of colistin resistance.
Conclusion:
The observed finding that points to a scenario of potential gut-associated resistance genes to Gram negative (K. pneumoniae) host in the NICU environment warrants attention and further investigation.
Background
Antimicrobial resistance among bacterial pathogens infecting patients in intensive care units (ICUs) remains a serious public health threat in the hospital setting. The acquisition of multidrug resistant (MDR) organisms in ICUs impose a significant socioeconomic burden on both individuals and the health care system.1,2 The clinical impact of MDR bacterial infections is of great concern in the neonatal intensive care unit (NICU) due to the risk factors and predisposing underlying conditions of the newborns, including but not limited to empirical antibiotic treatment regimes, cross-infection, exposure to broad spectrum antibiotics, and the use of invasive devices.3,4 Intestinal carriage of MDR Gram negative bacteria, in particular the extended spectrum β-lactamase (ESBL)–producing strains, has been shown to be an important risk factor for neonatal bloodstream infection. 5
In Asia, Klebsiella spp. have been identified as the most common bacterial pathogen causing late-onset sepsis in neonates.6,7 Previous studies have reported the presence of CTX-M-15 and its association with resistance to cephalosporins and aminoglycosides in the majority of the MDR Klebsiella pneumoniae isolates from Malaysia and other Asian countries.8,9 However, these MDR K. pneumoniae are typically sensitive to polymyxins. Based on the Malaysian National Surveillance of Antibiotic Resistance (NSAR), resistance rate of carbapenem in K. pneumoniae was less than 0.3% in 2011, 10 but resistance rate of imipenem and meropenem has increased almost 10 fold to 2.7% and 2.9%, respectively, in 2017. 11
In fact, an increasing trend of resistance to last resort antibiotics such as third generation cephalosporins and fluoroquinolones have been observed among the K. pneumoniae population in Malaysia.12,13 On the contrary, the rate of resistance to lipopeptide antibiotics such as polymyxin B and colistin among the K. pneumoniae populations in Malaysia may be under-represented. These antibiotics are not included in the groups of regularly tested antibiotics under the Ministry of Health surveillance system; hence, little or no data were available for the polymyxin resistance rates. 11
In addition, studies also have demonstrated that modifications of two-component regulatory systems, PmrAB and PhoPQ, and inactivation of the mgrB gene (a negative regulator of the PhoPQ signaling system) led to polymyxin resistance by alteration of the lipopolysaccharide target. 14 Following the discovery of plasmid-borne colistin-resistant gene mcr-1 by Liu et al., 15 a total of eight mcr-1-positive Escherichia coli of clinical and nonclinical origins were confirmed in Malaysia, suggesting the potential dissemination of the mcr-1 gene among the bacterial populations circulating in this region. 16
In Malaysia, colistin is commonly added into animal feed for prophylactic and growth-promoting purposes in livestock farming. 17 This practice may have encouraged the development of colistin resistance among zoonotic bacteria. Indeed, a recent study in Malaysia has reported the isolation of K. pneumoniae strains harboring blaMCR-1 gene from swine, 18 and majority of the colistin-resistant K. pneumoniae strains also harbored blaTEM gene (92.3%), blaSHV (69.23%), and blaCTXM-1 (38.46%). 18
While colistin-resistant K. pneumoniae has been reported worldwide, there is still lack of the related genomic report, especially from Malaysia. Hence, the aim of this study was to elucidate the genetic mechanisms of antibiotic resistance and virulence of the colistin-resistant K. pneumoniae isolated in Malaysia using whole genome sequencing (WGS).
Materials and Methods
Bacterial strains
We identified two colistin-resistant K. pneumoniae K36 and B13 with colistin minimal inhibitory concentration (MIC) of >256 μg/mL from our previous study. These isolates also demonstrated high resistance to the antibiotics tested, namely amoxicillin, ceftriaxone, ceftazidime, cefoxitin, aztreonam, meropenem, tobramycin, and ciprofloxacin. 19 Both isolates were isolated from stool specimens of preterm infants admitted to the NICU at day 7 and day 14 of life, respectively. The isolates were maintained on Luria-Bertani medium at 37°C.
DNA sequencing, assembly, and annotation
The genomic DNA was extracted using Qiagen DNA Mini Kit (Qiagen, Germany) and eluted with ultrapure water. Extracted DNA was subjected to purity check with NanoDrop (Thermo Scientific, MA) and quantified using Qubit fluorometer (Invitrogen; Thermo Scientific, MA). The DNA sequencing library was generated using KAPA Library Amplification Kit (Illumina, CA) and sequenced on MiSeq (Illumina) platform with 250 bp paired-end reads. Raw reads generated by the sequencer was paired, trimmed, and assembled de novo using IDBA-UD (version 1.0.9). 20 The protein-coding genes were first predicted using Prodigal. 21 Genome annotation was also performed using the NCBI Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP) utilizing the GeneMarkS+, Glimmer, and tRNAscan-SE tools simultaneously 22 and the RAST server. 23
Annotation of antibiotic resistance genes
The K. pneumoniae strains antibiotic resistance genes (ARGs) were annotated using the Resistance Gene Identifier (RGI) from the Comprehensive Antibiotic Resistance Database (CARD) 24 and ResFinder, 25 where strict criteria were chosen for prediction. The RGI prediction of ARGs is based on two detection model types as follows: Protein Homolog and Protein Variant SNP (single nucleotide polymorphism) models. Protein Homolog models detect antimicrobial resistant protein sequences based on BLASTp hits to the curated protein sequences in CARD. The default BLASTp cutoff point is an expect value (E) of e−30. 26 In contrast, SNP models perform a similar search, but secondarily screen query sequences for curated sets of genetic mutations that confer antibiotic resistance.
Detection of genetic alterations associated with colistin resistance
The presence of mutations in colistin resistance determinants, such as mgrB, pmrA, pmrB, phoP, and phoQ, was determined using the BLASTN and MUSCLE. Deduced protein sequences were translated using ExPASy, and PROVEAN 27 was used to predict alterations of biological functions of the proteins. The type of insertion elements (ISs) present in mgrB gene was also determined by IS Finder. 28
Mapping of virulence factors
Virulence-associated genes in the assembled genomes were identified using the virulence factor database (VFDB). 29 The protein sequences of annotated genes were aligned against experimentally verified VFDB protein sequences (Set B) (downloaded on May 14th, 2018) using BLASTp v2.7.1+. 30 A query read was strictly assigned with a minimum 90% identity with E-values <10e−5 and hit bitscore values higher than 90 to exclude distant homologs. The strict filtering criteria allowed analysis to focus only on nearly identical matches. If more than one virulence genes overlapped at the same locus in the genome, only the best-aligned virulence gene was taken into account.
Multilocus sequence typing classification and phylogeny of colistin-resistant K. pneumoniae
The allelic profiles based on the seven housekeeping genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) and sequence types (STs) of the K. pneumoniae strains K36 and B13 were assigned using the PubMLST database*. Phylogenetic relationship between the local strains and 46 colistin-resistant K. pneumoniae strains isolated from Asia-Pacific countries was predicted by concatenating the nucleotide sequences of the seven multilocus sequence typing (MLST) housekeeping genes. Background information of colistin-resistant K. pneumoniae strains from Asia-Pacific countries is included in Supplementary Data S1. This set of isolates was collected intended to estimate the discrimination of MLST among strains with no documented colistin resistance mechanism links. The maximum likelihood method was used to construct a phylogenetic tree with 1,000 bootstrap replications using MEGA6.06 (Molecular Evolutionary Genetics Analysis) software.31,32
Whole genome sequence phylogenetic analysis of global strains of K. pneumoniae
For WGS phylogenetic analysis, 28 representative complete genomes of clinical and environmental K. pneumoniae from 14 regions around the world (Malaysia, Thailand, China, India, Taiwan, Australia, Cameroon, United Kingdom, France, Germany, Greece, Netherlands, United States, and Canada) were included to determine their genetic relatedness with our K. pneumoniae strains. The names and respective NCBI GenBank accession numbers of the selected strains are as follows: HS11286 (CP003200), 1kgm (JXBI00000000), 223/14 (JRTV01000000), GN8 (NZ_JWPY00000000), FDAARGOS_440 (CP023919), BJ1-GA (CBTU000000000), SB3432 (FO203501), MGH155 (NGTB01000000), BWH67 (NGSR01000000), CAV1016 (CP0179341), DHQP1002001 (CP016811), CR14 (CP015392), Kp_Goe_821588 (CP018692), INF322 (CP024482), AUSMDU00008119 (CP025008), BR (CP015990), YH17175 (NZ_PTMB00000000), WCHKP1845 (MPOD00000000), KP36 (CP017385), QS17-0029 (CP024038), F1_9_ERG2 (NZ_FLYA00000000), HPCN17 (NZ_QGNV00000000), EGD-HP19-C (AUTW02000000), VBA2612 (PYSL01000000), PN030E4 (PDVM00000000), LKP723703-1 (PELJ00000000), M104145 (JTGA00000000), and ERS381189 (NZ_FLFQ00000000). Genome sequences were submitted to the Reference Sequence Alignment-based Phylogenetic Builder (RealPhy) 33 and aligned with default parameters of a 50-bp read length and 22-bp seed length 34 for the maximum likelihood method PhyML. 35 The K. pneumoniae HS11286 from China was chosen as the reference genome based on its highest genomic similarity with our strains. HS11286 is a MDR clinical isolate from human sputum with a complete genome sequence. 36 The multiple genome sequence alignments generated were subsequently used to construct phylogenetic trees, based on the approximate maximum likelihood method using FastTree 2.1.10. 37
Results
General features of the K. pneumoniae genomes
The major features of the sequenced K. pneumoniae K36 and B13 genomes are summarized in Table 1. The numbers of predicted protein-coding genes in both genomes were between 5,122 and 5,233. In addition, both genomes harbored pseudogenes. The genomes also differed in the number of rRNA operons, correlating with the number of tRNA genes. RAST annotations showed that most annotated genes in both genomes were involved in carbohydrate, amino acids and derivatives, cofactors, vitamins, prosthetic groups and pigment formation, protein, and RNA metabolism. Of these, 24 (K36) and 18 (B13) genes were associated with phages/prophages. There were also 132 and 147 potential virulence, disease, and defense related genes found in K36 and B13 genomes, respectively.
General Genome Features of the Sequenced Genomes
Association between antibiotic resistance phenotypes and genotypes
A total of 22 ARGs were identified in the genome of B13, while there were 19 ARGs in the K36 strain. Table 2 summarizes the antimicrobial resistance profiles and the corresponding ARGs identified in the genomes of K. pneumoniae K36 and B13 strains. Supplementary Data S2 shows detailed information of ARGs identified by CARD and ResFinder. Functional analysis of these ARGs revealed that B13 and K36 genomes harbored blaSHV-27 and blaSHV-71, respectively, contributing to the cephalosporin resistance phenotype of the strains. Detection of ompK37 gene in both K36 and B13 genomes was related to the resistance of certain β-lactams such as penicillins, cephalosporins, cephamycins, monobactams, and carbapenems. Based on the resistance mechanism categorized by CARD, B13 and K36 shared 13 ARGs related to antibiotic efflux, including oqxAB, emrR, adeF, and msbA. The floR and acrA genes were not detected in K36 while vgaC gene was not detected in B13; otherwise, they both had the same efflux pump complement.
Antimicrobial Resistance Profile and the Presence of Corresponding Antibiotic-Resistance Genes in the Klebsiella pneumoniae B13 and K36 Genomes Predicted by CARD and ResFinder
The R/I/S column provides information of the susceptibility to respective antibiotics: R = resistant, I = intermediate, S = sensitive.
AMC, amoxicillin/clavulanic acid; AML, amoxicillin; ARG, antibiotic-resistant gene; ATM, aztreonam; AZM, azithromycin; C, chloramphenicol; CAZ, ceftazidime; CIP, ciprofloxacin; CN, gentamicin; CRO, ceftriaxone; CT, colistin; FOX, cefoxitin; IMP, imipenem; MEM, meropenem; PB, polymyxin B; TOB, tobramycin; TZP, piperacillin/tazobactam.
Colistin resistance mechanisms
Annotation by CARD and ResFinder did not detect ARGs related to colistin resistance. To unravel the molecular mechanisms contributing to colistin resistance, the role of alterations of the MgrB and the two-component regulatory systems phoPQ and pmrAB were investigated. A base substitution mutation (R256G) of the PmrB-encoding gene was identified in K36, and the resulted PmrB variant (Arg256Gly) was considered deleterious by PROVEAN (Table 3). Both of the strains K36 and B13 exhibited alterations in the mgrB gene, including disruption by IS5708 (IS110 family). IS5708 is inserted within a putative 38-bp transposon TIR sequence, one side of the seventh base from the end of the transposon. In addition, deleterious mutations Leu17Gln and Gln22Leu were detected in MgrB (Fig. 1).

DNA and deduced protein sequence alignments of mgrB and pmrB alleles.
Mutations Reported Within Genes Involved in Colistin Resistance (mgrB, pmrAB, phoPQ)
WT, wild type.
Mutation predicted as deleterious by PROVEAN.
Virulence determinants of K. pneumoniae K36 and B13
The existence of 51 (K36) and 44 (B13) virulence-associated genes in the K. pneumoniae strains was determined in our study. VFDB assigned keywords for functional characterization were used for an overall categorization of the virulence genes (Table 4). Complete information of virulence-associated features of the K. pneumoniae strain B13 and K36 is shown in Supplementary Data S3. In general, both genomes of K. pneumoniae K36 and B13 shared almost similar virulence gene profiles, except that strain K36 had additional Ybt-like molecules, which were related to yersiniabactin siderophore production and iron regulatory proteins, which are responsible for iron uptake.
Klebsiella pneumoniae K36 and B13 Virulence Gene Homologs
VFDB, virulence factor database.
MLST phylogeny of colistin-resistant K. pneumoniae from Asia-Pacific regions
The maximum likelihood tree constructed from the seven housekeeping genes of the 48 colistin-resistant K. pneumoniae strains originated from Asia-Pacific countries were resolved into three main clusters (cluster I, II, and III) (Fig. 2). A total of 39 STs were distinguished. Our two Malaysian strains were grouped in cluster Ia and the strains and showed close phylogenetic relationship with majority of the strains from Laos.
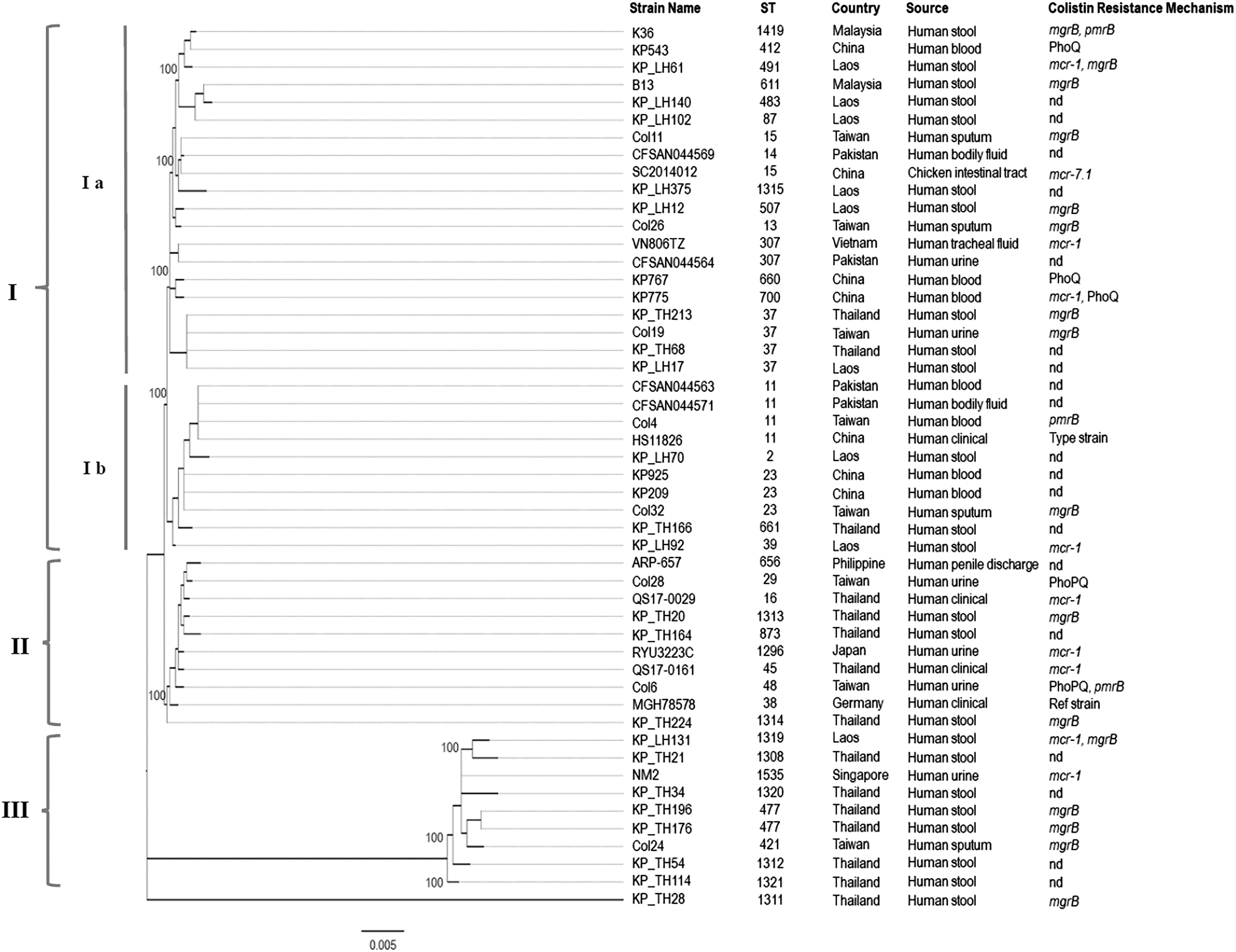
Maximum likelihood tree of MLST displaying the relatedness of the 48 Klebsiella pneumoniae isolates from countries of the Asia-Pacific region. The sequence types, countries, source of isolation, and colistin resistance mechanisms were indicated for comparison. The values on the nodes indicate the bootstrap value with 1,000 times resampling. MLST, multilocus sequence typing; nd, not detected.
Whole genome sequence phylogeny
The phylogenomic tree obtained from WGS is consistent with previous tree generated using MLST in which (1) strain K36 and B13 were clustered together, and (2) the clades and subclades in the phylogenetic trees were not defined geographically (Fig. 3).

Klebsiella pneumoniae core genome phylogeny inferred by approximately-maximum-likelihood method from the aligned core genomes. Multiple genome alignments were generated by mapping genome sequences of the 30 global K. pneumoniae strains with reference to strain HS11286 at all sites relevant for phylogenomic analysis using RealPhy. Phylogenetic trees in midpoint rooting was inferred using approximately-maximum-likelihood method using FastTreeMP. Bootstrap support values shown at each node. Scale bars indicate substitutions per site. Tips show strain name and source of isolation (host, geographic origin). Arrowheads indicate the strains of current study, and (★) indicates the reference strain.
Discussion
In the present study, we have investigated genetic factors, including mutations that underline the antibiotic-resistance phenotypes in the two clinical isolates of K. pneumoniae. Genome annotations of ARGs revealed the presence of a wide range of genes conferring resistance to major antibiotics. Both K36 and B13 genomes harbored oqxAB genes, which are responsible for fluoroquinolone resistance. Compatible with this notion, previous studies have shown that the resistance/nodulation/division superfamily multidrug efflux pump oqxAB was found to be conserved in all tested K. pneumoniae strains, thus further revealing an elevated and widespread dissemination of OqxAB efflux pump.38–40 Alteration in the antibiotic target sites is a common mechanism for MDR strains. In the current study, both K36 and B13 strains shared some common ARGs, which are responsible for antibiotic target alteration, such as marR and uhpT, as well as the mutations in penicillin-binding protein 3 (PBP3). Previous studies have shown that mutations in PBP3 related to antibiotic target alteration could confer higher resistance in amoxicillin, cefotaxime, and other β-lactams.41,42 In the current study, two amino acid mutations (S357N and D350N) in PBP3 were detected in both K36 and B13 genomes. These mutations were commonly found in clinically isolated Haemophilus influenzae with penicillins and cephalosporin resistance. 43 However, PBP binding data for K. pneumoniae are relatively lacking thus representing a significant knowledge gap. 44 Single amino acid polymorphism (E350Q) in UhpT was detected in genomes of K36 and B13. Mutation in uhpT related to fosfomycin resistance has been previously described. 45 There has been a resurgence in clinical use of fosfomycin because of its broad spectrum activity against many extensively drug-resistant Gram-negative bacteria.46,47 Therefore, high global prevalence of fosA genes that confer fosfomycin resistance warrants some attention. 48 Although we did not test for fosfomycin susceptibility of the strains examined in this study, FosA6 homolog was detected in both of the studied genomes. Phylogenetic analyses have revealed frequent lateral distribution of fosA6 from K. pneumoniae to other Gram-negative bacterial species, especially E. coli. 49 Consistent with this notion, detection of E. coli transporter protein UhpT homolog in our genomes shows that a reservoir of fosfomycin resistance determinants in the genomes of K. pneumoniae is transferrable to non-FosA-producing species. Presence of the florfenicol resistance gene floR in one of our human clinical isolates, B13, as well as animal and human isolates in another study from Malaysia, 16 highlighted the pressing need to curtail antibiotic usage in animal husbandry in the country.
A longitudinal study showed that the development of gut microbiome of two preterm infants admitted to the NICU was influenced by microbes in the NICU environment while the antibiotic-resistant microbes were colonizing these infants' gut. 50 A substantial number of ARGs encoding antibiotics are not being used in the NICU of current study, including polymyxins and chloramphenicol, suggesting that the bacterial resistome may have established at very early stage in life in other habitats, rather than direct antibiotic selection in the host or the environment (NICU). 51 Consistent with previous study, 52 we suggested that community acquisition of ARGs and maternal colonization of MDR isolates before hospital admission may be important in the neonatal population. These observations emphasized the importance and scale of the difficulty in mitigating the endemic spread of MDR K. pneumoniae. The process of gut-associated resistome suggested that the ARGs could be intertransferable among the gastrointestinal microbes, 53 especially potential pathogens during stress. 54 However, there are limited studies on the effect of gut colonization and gut-associated resistome transferability among preterm neonates over the NICU admission, suggesting that this knowledge gap should be filled to provide evidence for the clinical rise of pathogens.
The alteration of protein sequence, PmrB Arg256Gly, found in this study had also been reported by Aires et al., among the colistin-resistant isolates. 55 However, colistin resistance caused by this specific mutation alone was not significant as other secondary factor(s) may also be contributing to the resistance. 56 Inactivation of mgrB had also been demonstrated in colistin resistant isolates from different geographical locations such as Italy, 57 Spain, 58 France, Laos, Thailand, and Nigeria. 59 It was reported that the disruption of mgrB gene regulator could occur in different forms such as the truncation with various ISs, nonsense mutations leading to premature stop codons, or missense mutations. 59 The common mechanisms reported for mgrB disruption in K. pneumoniae were insertional inactivation by IS1-like, IS10-like, and IS5-like elements.55,60–62 In the current study, an insertion sequence, which belongs to IS110 family, has been identified in both strains. Such insertional inactivation due to IS pointing to be a source of colistin resistance in K. pneumoniae had been previously described by Poirel et al. 62 Further experiments are necessary to validate the finding.
Current gradual emergence of colistin resistance among the Gram-negative bacteria shows that clinical use of colistin may not be the only reason for such occurrence. Results obtained from previous studies showed that healthy individuals could also be colonized by colistin-resistant K. pneumoniae, and such high prevalence was observed in healthy individuals who had never received colistin therapy in rural areas of Southeast Asia.59,63 Zhang et al. reported identical nucleotide sequences of mcr genes of human and animal samples from the same city, suggesting the potential transmission of mobile colistin resistance determinants from animals to humans. 64 It was reported that the occurrence of colistin resistance among isolates from food animals is higher compared with human isolates, and such observation has been related to higher selective pressure due to injudicious use of colistin in animal husbandry. 65 Therefore, a “One Health” approach is important to integrally analyze colistin resistance in both humans and animals to combat the global rise of MDR microorganisms. Although our clinical isolates were not associated with infection in the host, K. pneumoniae is capable of silently colonizing hospitalized patients or hospital personnel through its inherent selective advantage in the hospital environment, where usage of antibiotics is widespread. 66 The organism may persistently reside in the gastrointestinal tract of the host without causing any signs of infection for a long period, thus making detection of these carriers difficult. 67 Silent intestinal carriage of MDR K. pneumoniae has been shown to be one of the important risk factors for continued transmission and nosocomial spread, especially in the ICU.68–70 Therefore, effective nosocomial outbreak control of K. pneumoniae requires a detailed understanding of the occurrence of transmission routes. Exploitation of molecular typing such as pulsed-field gel electrophoresis and MLST has been helpful to detect local and global dissemination. However, high clonality of K. pneumoniae limits the accuracy of the methods mentioned.68,71 Whole-genome sequencing technology is becoming more accessible with continuing improvements in turnaround time and cost and has been proven to be successful in tracking worldwide epidemics, 72 regional, 73 and institutional outbreaks. 74 Comprehensive sequence information provides evidence for unexpected transmission routes thus giving actionable insights in facilitating the control of nosocomial transmission. 75
In the current study, most of the virulence genes detected in both K36 and B13 genomes were associated with bacterial adherence mechanisms (15 genes), followed by genes associated with secretion and biofilm formation. Synthesis of yersiniabactin siderophore system in strain K36 suggested that there could be an increased risk for severe infections in humans, such as bacteremia, compared to strain B13. 76 Major fimbrial subunits encoded by the fimA and mrkA genes and a homolog of the E. coli common pilus (ECP) ecp operon, which are crucial for type 1 and type 3 pili production, were present in both of our K. pneumoniae genomes. Type 1 pili are the commonest adhesive organelles in Enterobacteriaceae, while type 3 pili are adhesins that mediate the binding of K. pneumoniae to endothelial and epithelial cells, leading to respiratory and urinary tract infections. 77 A PCR-based survey demonstrated a strong relationship between ecpA gene-bearing isolates and ECP production during adhesion and biofilm formation, suggesting that ECP may represent a new important adhesive structure for this organism. 78 Expression of bacterial pili, in particular ECP production, is under the influence of environmental signals such as oxidative stress. Although the underlying molecular mechanisms are not fully understood, this finding has led to the discovery of ECP as a biological marker for their ability to colonize the host intestinal epithelium.79,80 Biofilm forming communities and efflux pumps are conferred competitive advantages in many Enterobacteriaceae, contributing to their adhesion to the host epithelial cells and succession in the gut. 81 In addition, detection of FimDHGFC complex in our K. pneumoniae genomes indicated the presence of the chaperone-usher pathway, which is responsible for pili secretion to mediate host cell recognition and adherence. Some consideration has to be given to the interplay between bacterial response to its immediate environment and protein secretion because the ability to secrete proteins extracellularly has long been recognized as a major and exploitable feature of Gram-negative bacteria. 82 An array of pullulanase secretion genes (pulBDEFGHIJKLNOS) was identified in both genomes of K36 and B13. Pullulanase is a lipoprotein secreted to the surface of Klebsiella spp. through the type 2 secretion system, which is important for hydrolysis of polysaccharides and immune evasion but does not contribute directly to bacterial virulence in vivo.83,84 The finding in this study proposed that the pathogenicity of our strains could be reliant on dedicated secretion systems to transport virulence proteins outside the cell or target cell as described by Francetic and Pugsley. 85 Generally, virulence genotypes and phenotypes in relation to antimicrobial resistance profile are poorly studied in K. pneumoniae that cause nosocomial infections. However, a correlation between ESBL production and some virulence factors has been proposed. 86 The use of Galleria mellonella infection assays to study microbial virulence factors for K. pneumoniae has only recently been demonstrated. 87 Esposito et al. demonstrated the heterogeneity of the virulence phenotypes and genotypes, yet the level of colistin MIC of K. pneumoniae could predict its infectivity capability in G. mellonella. 88 With the genomic data provided in this study, more targeted experiments could be performed to determine the complex relationship between the heritable virulence genetic traits and the actual physical characteristics causing symptoms in the host, as well as the correlation with reduced susceptibility to antimicrobial agents.
Occurrence of MDR K. pneumoniae with colistin resistant phenotype has been reported within the Asia Pacific regions; however, its epidemiology and endemic transmission are poorly understood. There is no extensive study available to support the phylogeographic relatedness between Malaysian and Laotian strains. Strains from China showed limited genetic variation as all strains were grouped in cluster I. On the contrary, relatively greater diversity was observed among K. pneumoniae from Thailand and Taiwan, in which the strains were found distributed in all three clusters. The phylogenetic clades generated from MLST scheme, which utilizes conserved housekeeping genes, were not defined by the mutation/inactivation and plasmid-bearing genotypes. This highlights the limitation of MLST for clustering highly recombinogenic strains. 89 This observation concurred with previous study in which isolates with colistin-resistant phenotype were not restricted to a particular ST, instead they were diverse in clonal background. 90 In the current study, K. pneumoniae isolates with colistin resistance mechanism related to mgrB mutations were discriminated into 13 STs. Previous studies reported that isolates belonging to ST11 and ST258 showed independent events of mgrB gene disruption.55,56,62 ST11 was the most commonly detected ST among the colistin-resistant K. pneumoniae; such phenomenon has been proposed to be correlated with the strain capsular type instead of the seven-gene MLST scheme.56,91 As previously reported, whole genome phylogeny provided higher resolution of strain differences with better support of internal branches. 92 K. pneumoniae K36 showed closest phylogenetic relationship to a Malaysian strain isolated from water collected from a farm, GN8; however, this study has not been published. The close phylogenetic relationship among the K. pneumoniae strains isolated from humans and aquatic environment is not an uncommon phenomenon. Previous study done has shown that water bodies such as lakes, waste water, and streams appear to play crucial role in the emergence and dissemination of antimicrobial-resistant organisms, thus highlighting the pressing need to improve water quality worldwide. 93 The K. pneumoniae strains in Malaysia were genetically diverse and constitute the major clades and two other minor clades. There was no correlation observed between the sources of isolation and geographical origins of the K. pneumoniae strains. For instance, strains PN030E4, KP36, YH17175, and MGH155 essentially shared the core genome under the same cluster while originating from Cameroon, Taiwan, China, and the United States, respectively, and were obtained from either humans or poultries. In addition, a case of K. pneumoniae isolated from a farm in Thailand was grouped in the same subclade with human isolates from the United States, Australia, and Netherlands, suggesting possible dissemination of this pathogen between animals and human. The epidemiology of our Malaysian strains provided evidence suggesting that environmental contamination may play a role in transmission.
Conclusion
Our data provided detailed genetic background and revealed the presence of an array of antibiotic resistance and virulence genes pertaining to colistin-resistant MDR K. pneumoniae, which contributed to the bacterial persistence and pathogenicity. In addition, disruption of mgrB gene was shown to be associated with colistin resistance in the K. pneumoniae strains. The finding also suggests the potential of gut-associated bacterial resistome establishment during very early stage of life from other habitats, thus representing an example of infectious disease threat that requires the One Health approach. As the application of genome sequencing to study clinical pathogens grows rapidly, our analyses promote a deeper appreciation of the genetic diversity and epidemiologic data of this important pathogen and, therefore, provide further insights into the factors underlying the clinical rise of MDR K. pneumoniae.
Declarations
Ethics approval and consent to participate
The study has been approved by the University of Malaya Research Ethics Committee (UMREC) with ethical approval number 201310-0267. Informed consent was obtained from all study subjects.
Availability of data and material
The whole genome shotgun projects for strains K36 and B13 have been deposited in DDBJ/EMBL/GenBank under the accession numbers PXHX00000000 and PXYF00000000, respectively. The versions described in this article are the first versions.
Footnotes
Authors' Contributions
C.S.J.T. designed the experiments, selected the isolates, funded the project, and approved the article; A.A.K. assisted in the study design and helped obtain the isolates. P.S.X.Y. collected the samples, obtained the isolates, performed the experiments and analyses, as well as wrote the article. C.W.C. and S.T.N. suggested and commented on the study design and helped write the article. All authors read and approved the final article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by University of Malaya Research Grant (UMRG: RG353-13HTM) and Postgraduate Research Funding (PPP: PG179-2015A).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.