
Abstract
Clostridioides difficile ST37 is an emerging and prevalent multilocus sequence type and represents a lineage of clinical significance. This study aimed to characterize two epidemic C. difficile ST37 strains, CD161 and CDT4. CD161 acquires a chromosome and two distinct plasmids, pCD161-L, sharing high similarity with Clostridium phage, and pCD161-S, while CDT4 has a chromosome and a plasmid pCDT4 identical to pCD161-S. In the chromosome of both strains, three CdISt1-like elements and a skinCd element, which might influence sporulation, were identified. The multidrug resistance of the strains was due to the mutation in 23S rDNA, gyrA, and gyrB genes and the acquisition of ermB, ant6-Ia, aac6’-aph2’’, and tetM genes. In addition, a distinct pathogenicity locus (PaLoc) with truncated tcdA gene represents the genetic feature of ST37 strains. To our knowledge, this is the first complete genome, both chromosomes and plasmids, of epidemic C. difficile ST37 strains in China.
Introduction
C
Recently, the pathogenic A-negative B-positive (A− B+) strains (which produce TcdB, but not TcdA) have appeared and become prevalent in Asia (Hawkey et al.). 3 ST37, ribotype 017 (RT017) strains, which were the second A− B+ group to be identified, represent a lineage of clinical significance 4 since they have been responsible for CDI outbreaks in many countries, including the United States, Ireland, Netherlands, Germany, and China.3–5 A systematic analysis on tcdA and tcdB in C. difficile had reported that ST37 has low genetic diversity and shares connected features in its tcdA and tcdB genes with some known hypervirulent strains, highlights the potential epidemic threat of ST37 in China. 5
Despite a total of 52 complete genome and 11 plasmid sequences of C. difficile being available at NCBI up to date (excluding CD161 and CDT4), however, only 3 C. difficile strains (M68 from Ireland, CF5 from Belgium, and DSM 29627 from Indonesia) with no plasmid reported belong to ST37, suggesting insufficient genomic information for ST37 strains. Thus, in this study, the complete genome of two ST37 C. difficile strains isolated in China was sequenced and analyzed, to provide information on limited genomic features of epidemic ST37 strains.
Materials and Methods
Bacterial strains
A total of 467 stool samples were collected from diarrhea patients in the First Affiliated Hospital of Guangzhou Medical University in Guangzhou, China, in 2013. Bacterial identifications were performed by Gram staining and PCR amplification targeting tpi, a species-specific housekeeping gene C. difficile. 6 Among, 22 samples tested positive for C. difficile. C. difficile isolates CDT4 and CD161 were isolated from the stool samples of two individual diarrhea patients and subjected to further study.
Antibiotic susceptibility testing
Minimum inhibitory concentration (MIC) testing of 10 antibiotics (metronidazole, vancomycin, ampicillin, clindamycin, moxifloxacin, meropenem, amoxicillin/clavulanate, piperacillin/tazobactam, penicillin, and piperacillin) was performed by Etest (AB bioMérieux, France) and further interpreted according to the Clinical and Laboratory Standards Institution guidelines (CLSI, 2016) for C. difficile and standard instructions of Etest™ strips.7,8 C. difficile ATCC700057 was used as a control.
Multilocus sequence typing
Total DNA of the two isolates was extracted using a bacterial DNA extraction kit (Sigma-Aldrich) and quantified by pulsed-field gel electrophoresis and Qubit dsDNA BR assay (Life Technologies). The seven housekeeping genes (adk, atpA, dxr, glyA, recA, soda, and tpi) were amplified and the PCR products were purified and sequenced with ABI Prism 377 DNA Sequencer (PE Applied Biosystems). DNA sequences were submitted to PubMLST to obtain the sequence types (STs), which were also confirmed using their genome sequences to query the PubMLST using MLST 2.0. 9 The STs of the total genome-sequenced C. difficile strains were also identified using their genome sequences available in GenBank database through MLST 2.0.
Genome sequencing and assembly
Genomic DNA of the two isolates was sequenced using Illumina HiSeq 4000 and PacBio RS II sequencers. Low-quality data were filtered before further assembly. Four parts of assembly, including subread correction, corrected read assembly, single-base correction, and sequence loop judgment and chromosome, plasmid, sequence discrimination, were performed. 10 The Pbdagcon program was used for subread correction. Corrected reads were assembled using the Celera Assembler against a high-quality corrected circular consensus sequence subread set. To improve the accuracy of the genome sequences, GATK and SOAP tool packages (SOAP2, SOAPsnp, SOAPindel) were used to make single-base corrections. To trace the presence of plasmids, the filtered Illumina reads were mapped using SOAP to the bacterial plasmid database.
Genome functional annotation
Genes were predicted by Glimmer (Version:3.02) with hidden Markov models and further annotated by blasting genes with NR database, respectively. The tRNAs, rRNAs, and sRNAs were identified using tRNAscan-SE, RNAmmer, and the Rfam database, respectively. Prophage regions were predicted using the PHAge Search Tool web server. Antibiotic resistance genes were predicted through Antibiotic Resistance Genes Database and ResFinder 3.0, 11 and virulence factors were predicted by Virulence Factors of Pathogenic Bacteria database.
Nucleotide sequence accession numbers
The complete sequences of C. difficile strain CDT4 have been deposited in GenBank under accession numbers CP029152 (chromosome) and CP029153 (plasmid pCDT4). The complete sequences of C. difficile strain CD161 have been deposited in GenBank under accession numbers CP029154 (chromosome), CP029155 (plasmid pCD161-L), and CP029156 (plasmid pCD161-S).
Ethics statement
All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by Medical Ethics Committee of the First Affiliated Hospital of Guangzhou Medical University. Informed consent was obtained from all subjects.
Results
General features of the bacterial and genome sequences
Clostridioides difficile strains CDT4 and CD161 are multidrug-resistant isolates showing resistance to clindamycin, moxifloxacin, and penicillin, and belong to ST37 (adk3, atpA7, dxr3, glyA8, recA6, soda9, and tpi11). Whole-genome sequencing and assembly yield a 4,295,210 bp chromosome, a 130,529 bp plasmid pCD161-L and a 48,594 bp plasmid pCD161-S with an average GC content of 28.85% in CD161 and a 4,240,954 bp chromosome and a 48,594 bp plasmid pCDT4 with an average GC content of 28.88% in CDT4. In the genome of CD161, 4137 coding sequences (CDS), 90 rRNAs, 35 tRNAs, 21 sRNAs, 607 tandem repeats, 470 minisatellite DNAs, and 20 microsatellite DNAs, were identified. In the genome of CDT4, 3887 protein-CDS, 89 rRNAs, 35 tRNAs, 21 sRNAs, 607 tandem repeats, 470 minisatellite DNAs, and 20 microsatellite DNAs were identified. Besides, 12 and 2 credible CRISPR were predicted in the CD161 chromosome and plasmid pCD161-L, respectively, while 12 credible CRISPR were acquired by the chromosome of CDT4.
Comparative genome analysis
The chromosome of CD161 showed 99% identity and 97%, 97%, 96%, and 96% coverage with those of CDT4 and other three completely sequenced ST37 C. difficile strains, M68 (FN668375), CF5 (FN665652), and DSM 29627 (CP012325), respectively (Fig. 1A). Particularly, a distinct 71.4 kb prophage was identified in the chromosome of CD161. The plasmid pCD161-S backbone is identical to pCDT4 and partial sequence of M68 chromosome (4254330–4302721 bp), which might be an individual plasmid (Fig. 1B). It also showed 84% coverage and 98% identity to the plasmid sequences of ST3 C. difficile strains AK (pAK2, CP027016), FDAARGOS_267 (unnamed1, CP020425), and ATCC 9689 (unnamed, CP011969) (Fig. 1B). The larger plasmid pCD161-L backbone showed 69% coverage and 97% identity to the plasmid sequences of C. difficile strains AK (pAK1, CP027015) and FDAARGOS_267 (unnamed2, CP020426), and Clostridium phage phiCD211 (LN681537) (Fig. 1C).
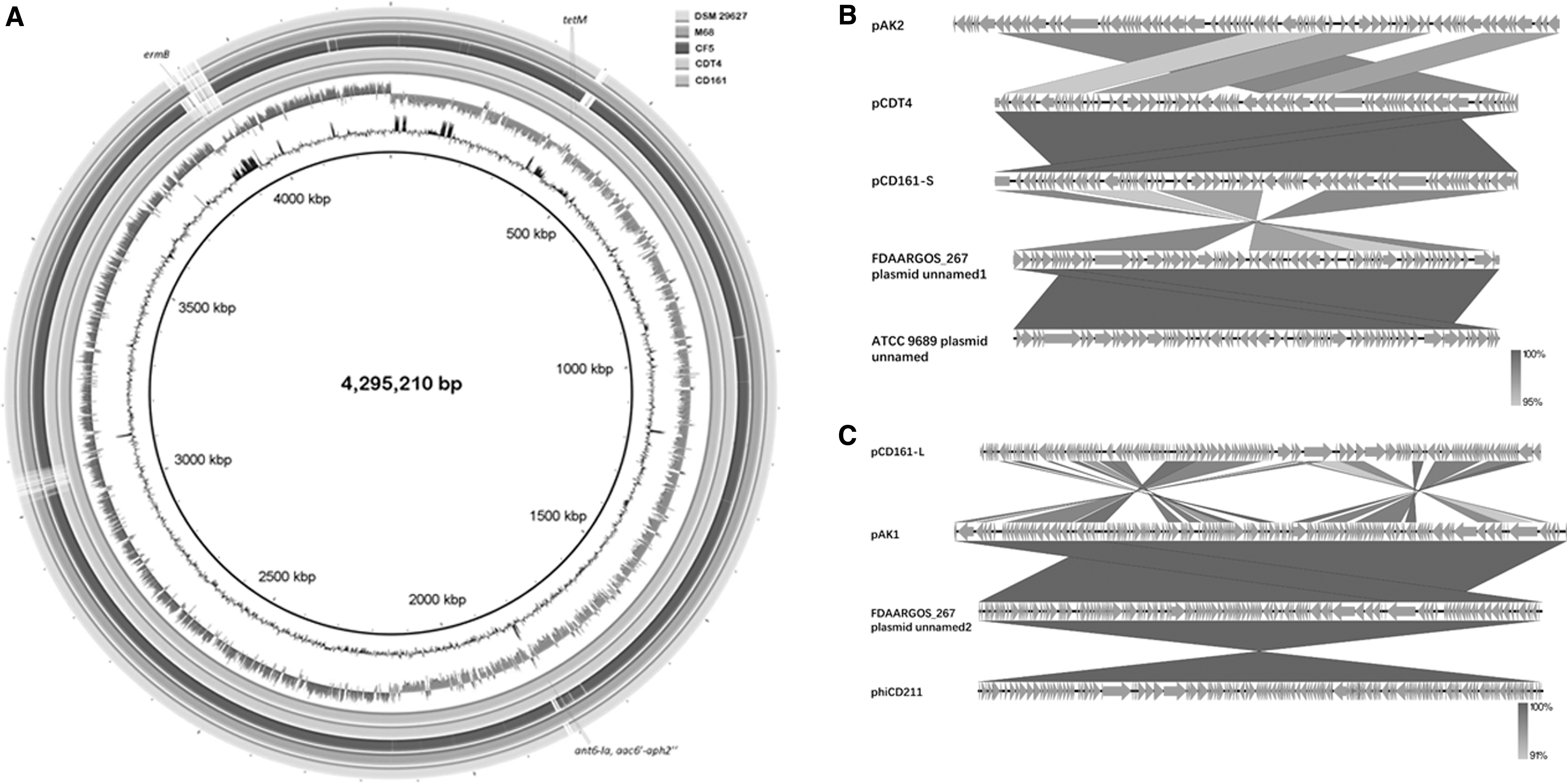
Schematic map of Clostridioides difficile CD161 chromosome
Antibiotic resistance genes
Antibiotic resistance genes ant6-Ia, aac6′-aph2″, and tetM were identified in both chromosomes of CD161 and CDT4. The T82I (ACT-ATT) mutation in the gyrA gene and S366A (TCA-GCA) mutation in the gyrB gene were also acquired by both chromosomes of CD161 and CDT4. The acquisition of an ermB gene was specifically identified in the chromosome of CD161. A 23S rDNA mutation with a nucleotide substitution (1962 C→T) was identified only in CDT4.
Mobile genetic elements and virulence factors
In the chromosome of C. difficile strains CD161 and CDT4, three CdISt1-like elements and a 16.7 kb C. difficile sigK intervening sequence (skinCd) element were identified. The CdISt1-like elements in C. difficile strains CD161 and CDT4 were identical to CdISt1 but not located within the tcdA gene. The 16.7 kb skinCd element was found to interrupt the sigK gene.
The main virulent factors of C. difficile are toxin A and toxin B encoded by tcdA and tcdB genes, respectively. Specifically, the tcdA gene in C. difficile strains CD161 and CDT4 was truncated (Fig. 2), consistent with the A− B+ features of ST37 strains.

Schematic map of PaLoc of Clostridioides difficile strains CD161 and CDT4 aligned with C. difficile strain 630. PaLoc, pathogenicity locus.
Discussion
Among the 22 C. difficile isolates identified from 467 stool samples, C. difficile strains CDT4 and CD161 are the only multidrug-resistant isolates showing resistance to clindamycin, moxifloxacin, and penicillin and belong to ST37, the dominant ST in China. 12 C. difficile ST37 strains have caused widespread disease across the world and showed persisting predominance in Asia. 13 Thus, whole-genome sequencing and assembly were conducted on the two C. difficile strains to better understand their pathogenic features.
The high chromosomal identity with other strains that belonged to ST37 implied the conserved chromosome in C. difficile ST37 strains. The similarity in plasmid sequences of ST37 strains (CD161, CDT4, and M68) and ST3 strains (AK and FDAARGOS_267) indicated the relatively close genetic connection between ST37 and ST3, the most prevalent STs in mainland China. 12 Besides Clostridium phage phiCD211, plasmid pCD161-L also showed relatively high similarity with phages LIBA6276 (MF547662), LIBA2945 (MF547663), and phiCDIF1296T (CP011970), indicating the phage-like genomic features of pCD161-L. Capable of reducing bacterial numbers in both in vitro and in vivo systems, phage therapy is an attractive treatment option for CDI. 12 A series of phage-related genes associated with tail morphogenesis (tail protein, tail tape measure protein, tail fiber domain-containing protein, etc.), DNA replication, modification, and metabolism (nuclease, DNA ligase, methylase, guanylate kinase, etc.) and host cell lysis (N-acetylmuramoyl-L-alanine amidase, etc.) were identified in pCD161-L.
CdISt1, a DNA insertion of 1975 bp, was first identified within the enterotoxin gene (tcdA) of C. difficile strain C34. 14 CdISt1 combines features of two genetic elements, the invasiveness of an insertion element and the splicing ability of a group I intron, rendering transposition harmless for the interrupted gene and may constitute a novel group of ribozymes that are specifically adapted to survive and spread in eubacterial genomes. 14 The CdISt1-like elements in C. difficile strains CD161 and CDT4 were identical to CdISt1 but not located within the tcdA gene, probably due to the truncation of tcdA gene in ST37 strains. The 16.7 kb skinCd element was found to interrupt the sigK gene, which encodes an RNA polymerase sigma factor essential for sporulation. 15 The 14.6 kb skinCd element in C. difficile strain 630 is excised from the mother cell chromosome only during sporulation and is required for efficient sporulation. 16 Compared with C. difficile strain 630, the skinCd elements identified in this study acquired two extra ORFs encoding the transcriptional regulator and hypothetical protein, and might have a similar function.
Clostridioides difficile CD161 and CDT4 are multidrug resistance isolates presenting strong resistance to clindamycin and moxifloxacin, and to a lesser extent, penicillin (Table 1). Accordingly, the T82I (ACT-ATT) mutation in the gyrA gene and S366A (TCA-GCA) mutation in the gyrB gene in both chromosomes of CD161 and CDT4 contribute to the resistance to moxifloxacin. 16 High-level resistance against macrolide/lincosamide/streptogramin (MLS) antimicrobials usually requires an erm gene, 17 and MLS resistance in C. difficile is encoded by the ermB gene located on a mobilizable conjugative transposon Tn5398. 18 The acquisition of an ermB gene by the chromosome of CD161 explained its clindamycin resistance. However, transposon Tn5398 was not identified, indicating a new genetic structure of ermB determinant. In addition, 23S rDNA mutation with a nucleotide substitution (C→T) in clindamycin-resistant but erm-negative isolates had been identified. 19 Thus, the 23S rDNA mutation in the erm-negative strain CDT4 might contribute to its clindamycin resistance. In addition, the antibiotic resistance genes ant6-Ia, aac6’-aph2’’, and tetM identified in both chromosomes of CD161 and CDT4 indicated their potential resistance to aminoglycoside and tetracycline.
Antimicrobial Resistance Pattern and Resistance Genes Identified in Clostridioides difficile Strains CDT4 and CD161
MIC, minimum inhibitory concentration; R, resistant; S, sensitive.
The low genetic diversity and the possible international spread of ST37 strains had been revealed in a systematic analysis of tcdA and tcdB in C. difficile due to the same sequences of tcdA and tcdB acquired by ST37 isolates from different regions. 5 The PaLoc in C. difficile strains CD161 and CDT4 is identical to other ST37 strains (M68, CF5, and DSM 29627), which verified the low genetic diversity. Except the truncation in tcdA gene, the tcdB, tcdR, tcdE, and tcdC genes are identical to those in A+ B+ strain 630, indicating the evolution path (deletion of the 3′ region of the tcdA gene) of ST37 strains from A+ B+ strains and the potential threat of ST37 strains. Concerning binary toxins, although cdtA and cdtB genes had been detected in several ST37 strains by PCR, both cdt genes were not identified in C. difficile strains CD161 and CDT4. 20 With different isolation sources, locations, and antibiotic susceptibility profiles, the virulence gene acquisition might differ. The defect of C. difficile strains CD161 and CDT4 in binary toxins might due to some mutations during evolution.
Conclusion
In conclusion, two multidrug-resistant C. difficile ST37 strains were completely sequenced and yielded a chromosome and two distinct plasmids (pCD161-L and pCD161-S) in CD161 and a chromosome and a plasmid (pCDT4) in CDT4. It represents the first complete sequence, both chromosomes and plasmids, of epidemic C. difficile ST37 strains in China. Specifically, pCD161-L shares high similarity with Clostridium phage. In the chromosomes, three CdISt1-like elements and a skinCd element, which might influence sporulation, were identified. The mutation in 23S rDNA, gyrA, and gyrB genes and the acquisition of ermB, ant6-Ia, aac6′-aph2″, and tetM genes contribute to the multidrug resistance. In addition, a distinct PaLoc with truncated tcdA gene representing the genetic feature of ST37 strains was identified. This finding may offer significant insight on the epidemiology of C. difficile ST37 strains.
Footnotes
Authors' Contributions
Z.X. and A.W. conceived the study and participated in its design and coordination, H.S. carried out strain collection and sample preparation. H.T. and L.P. conducted strain identification and antimicrobial susceptibility testing. J.L. and D.C. conducted DNA extraction, genome sequencing, assembly, and bioinformatic analyses. All authors reviewed and approved the final article.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Scientific and Technological Program of Guangzhou City (201510010241) and the 111 Project (B17018).