
Abstract
To reduce the high rates of morbidity and mortality caused by methicillin-resistant Staphylococcus aureus (MRSA) strains, it is essential to prevent their transmission. This can be achieved through molecular surveillance of the infecting strains, for which the detection of the entry of new strains, the analysis of antimicrobial resistance, and their containment are essential. In this study, we have analyzed 190 MRSA isolates obtained at the Consorcio Hospital General Universitario de Valencia (Spain) from 2013 to 2018 with three approaches: Multilocus Sequence Typing, spa, and SCCmec typing. Although the incidence of S. aureus infections detected in the hospital increased in the study period, the frequency of MRSA isolates decreased from 33% to 18%. One hundred seventy-two MRSA isolates were resistant to three or more classes of antimicrobials, especially to fluoroquinolones. No relevant temporal trend in the distribution of antibiotic susceptibility was observed. The combination of the three typing schemes allowed the identification of 74 different clones, of which the combination ST125-t067-IV was the most abundant in the study (27 cases). Members of three clonal complexes, CC5, CC8, and CC22, comprised 91% of the isolates, and included 32 STs and 32 spa types. The emergence of low incidence strains throughout the study period and a large number of isolates resistant to different classes of antibiotics shows the need for epidemiological surveillance of this pathogen. Our study demonstrates that epidemiological and molecular surveillance is a powerful tool to detect the emergence of clinically important MRSA clones.
Introduction
Methicillin-resistant S. aureus (MRSA) is one of the most important nosocomial pathogens due to the acquisition of resistance to several first-line antibiotics, 1 limiting the effective treatment options. 2 MRSA has been described as endemic in many hospitals; however, major epidemic outbreaks have also been reported. 3 This pathogen is responsible for almost 150,000 infections every year in EU and EEA countries. 4 Consequently, more than 7,000 deaths a year are attributed to this pathogen. 5 In 2017, WHO classified MRSA in the high-priority category for which new antibiotics are urgently needed. 6
Resistance to methicillin in MRSA is determined by the acquisition of the Staphylococcal Cassette Chromosome mec (SCCmec), which contains the genes mecA or mecC 7 producing resistance to methicillin. In addition, the SCCmec contains a complex of genes responsible for the mobility of the cassette, such as ccrA and ccrB. 3 Five main types of SCCmec (I-V) have been identified as well as multiple variants of them. They are classified according to the ccr and mec genes. 8 The larger cassettes (types I, II, and III) are strongly associated with the multidrug resistance phenotype9,10 and they have been found mainly in Health care-Associated MRSA (HA-MRSA) isolates. The smaller cassettes (types IV and V) have fewer resistance genes, but greater mobility and are related to Community-Associated MRSA (CA-MRSA) isolates, but also to widespread HA-MRSA clones. 7 However, the distinction between the two epidemiological groups, HA-MRSA and CA-MRSA, has become more diffuse. 11
According to the ECDC, 23.3% of the S. aureus isolates obtained in Spain in 2020 (2,292 isolates) were MRSA, which places Spain among the countries with the highest proportion of MRSA isolates in Europe. 12 This has reinforced the need to establish adequate epidemiological and genomic surveillance systems to prevent and control emerging outbreaks. 13
Epidemiological studies using molecular typing have proven essential in the determination of the genetic diversity and relationships of S. aureus isolates, as well as to track the spread of infections of the pathogen. 14 Currently, the most widely used typing methods are Multilocus Sequence Typing (MLST), 15 spa typing, 16 SCCmec typing, 8 and whole-genome sequencing, whose relevance has substantially increased recently. 17
MLST is a very useful method due to its high reproducibility and discriminatory power. Nevertheless, in those outbreaks in which endemic clones cannot be differentiated by MLST, spa typing is especially useful. 18 In addition, determining the SCCmec type allows a better discrimination within clonal complexes and inferring the hospital or community association of the variant studied and its antibiotic resistance profile. 8
This study aims to characterize the MRSA population over a 6-year period at the Consorcio Hospital General Universitario de Valencia (CHGUV), as well as to obtain a detailed image of its population characteristics and dynamics. For this, the MLST, spa, and SCCmec typing methods were combined for a detailed characterization of the variants isolated in the hospital, performing a phylogenetic analysis to determine their evolutionary relationships.
Materials and Methods
Sample selection
Identification and antimicrobial susceptibility testing of S. aureus strains were performed by MicroScan instrument using the Panel Type PC 31 (Beckman Coulter) following the Clinical and Laboratory Standards Institute recommendations (CLSI) guidelines of the year of strain isolation. 19
MRSA confirmation was performed by Clearview® PBP2a SA assay (Abbott) to detect penicillin-binding protein 2a (PBP2a) related to mecA gene carriage. If PBP2a tested negative, a commercial LAMP-PCR was performed (Eazyplex® MRSA, Amplex) to discard mecC production.
The first 30–35 MRSA strains isolated each year from 2013 to 2018 were selected for analysis. To avoid repeating isolates, only the first isolate from each patient was included in the sampling. No criteria regarding clinical, antimicrobial, or genetic aspects were applied for selection to study the population of this pathogen in the hospital. Relevant clinical data (date of specimen collection, specimen type, ward, patient age, gender, and the antimicrobial susceptibility pattern) were extracted from the laboratory and hospital records for all staphylococcal infections between 2013 and 2018. Community-acquired (CA) infections were defined when a MRSA was isolated from an outpatient or within 48 hrs of current admission as an inpatient. After 48 hrs of admission, patients were considered to have a health care-associated (HA) infection. Samples were collected under a surveillance program for antimicrobial resistance and informed consent was waived by the CEI of FISABIO-DGSP.
DNA extraction and quantification
The DNA of the selected isolates was extracted by heat shock (Supplementary Data S1). The nucleic acid concentration in each of the study samples was quantified using the NanoDrop™ 3300 (ThermoFisher Scientific) following the manufacturer's instructions. Aliquots with a concentration of 50 ng/μL of DNA in a 20 μL final volume were prepared for further analysis.
MLST and spa typing
The MLST and spa typing were performed according to Enright et al 15 and Frénay et al, 16 respectively, and are detailed in Supplementary Data S1. PCR reactions were performed using an Eppendorf™ Mastercycler. PCR products were purified by ultrafiltration using NucleoFast purification plates and following the manufacturer's protocol.
Sanger sequencing of the amplified regions was performed using the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). Sequencing primers were the same used in PCR amplification (Supplementary Table S1). The sequencing reactions were carried out in an Eppendorf Mastercycler. Capillary electrophoresis was performed at the Sequencing Service of the University of Valencia (SCSIE) using an ABI 3730XL DNA sequencer.
The chromatograms were analyzed with the Staden package 20 for assembling and obtaining the consensus sequence. STs were determined using the S. aureus PubMLST database. 21 The spa type was assigned using the Ridom SpaServer 22 and the online server SeqTools. 23
SCCmec typing
Among the different SCCmec typing methods, we followed the multiplex PCR strategy described by Milheiriço and Oliveira DC 24 as this is the most widely used. 18 Details of the procedure are indicated in Supplementary Data S1 and Supplementary Table S2. Electrophoresis was carried out in agarose gels at a concentration of 3% and a current of 4 V/cm for 2.5 hrs. The observed band patterns were compared with those described 24 to determine the SCCmec type.
Phylogenetic study and epidemiological analysis
To better understand the MRSA population at the CHGUV and to compare it with the Spanish population, two phylogenetic trees were obtained. The first tree includes only MRSA isolates from the CHGUV. The second tree incorporates 257 ST sequences deposited in the PubMLST database corresponding to MRSA and methicillin-susceptible S. aureus (MSSA) isolates from different Spanish locations (accessed on 20/05/2021).
Phylogenetic reconstructions were obtained from the concatenated sequences of the seven genes of the MLST scheme. Before concatenation, we aligned the sequences using Aliview and MUSCLE. 25 The evolutionary model was selected with MEGAX, 26 based on the Bayesian Information Criterion. 27 The maximum likelihood phylogenetic tree was generated using IQTREE 28 with 1,000 bootstrap replicates.
Finally, we used iTOL 29 to incorporate the results of the spa and SCCmec typing as well as the epidemiological data.
Results
Setting and S. aureus epidemiology
Between 2015 and 2018, the number of S. aureus cases increased in the hospital from 1,185 to 1,483, respectively. Although the number of MRSA cases accounts for a significant fraction of the total number of S. aureus cases, the trend decreased over the 5 years of study, falling from 33% to 19% between 2013 and 2018. MRSA dynamics remained uniform throughout each year. No seasonal peak, antimicrobial susceptibility variation, or hospital ward accumulation was reported outside the sampling periods. Selected strains represented about 10% of the MRSA yearly incidence at the hospital.
Sample collection and antibiotic susceptibility
We collected 190 samples from wound exudates (27.2%), ulcers (18%), nasal exudates (11.3%), urine (10.8%), sputum (8.7%), blood (7.2%), biopsy (6%), and others (10.8%). The mean age of the patients was 67 years [11 months–94 years], with a slightly higher proportion occurring in men (61.8%) than women. Forty-eight samples were considered HA-MRSA, whereas 142 corresponded to CA-MRSA.
In addition to beta-lactams, which include methicillin, 81% of the 190 MRSA isolates were resistant to three or more classes of antimicrobials, especially to fluoroquinolones (172 isolates), and aminoglycosides (106 isolates) (Supplementary Table S3). We found resistant strains to relevant antibiotics for the treatment of severe and skin infections, bacteremia, or right-sided endocarditis such as clindamycin (54, 28.4%), daptomycin (6, 3.2%), and trimethoprim/sulfamethoxazole (3, 1.5%) (Fig. 1). However, all the isolates were susceptible to linezolid, vancomycin, and teicoplanin. In concordance with the global trends for MRSA isolates (see S. aureus epidemiology), no relevant temporal trend in the distribution of antibiotic susceptibility was observed in the studied isolates.
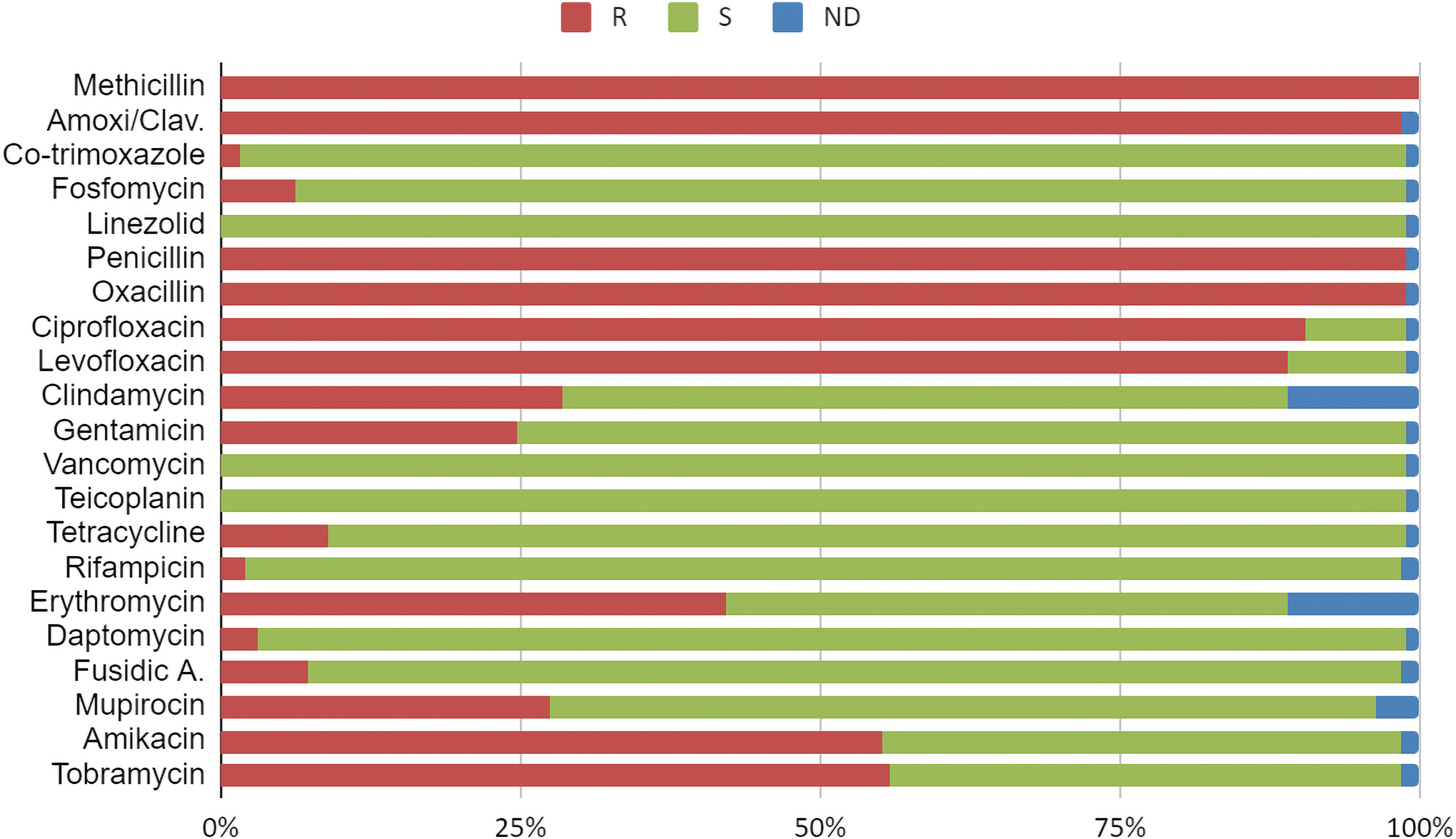
Proportion of the 190 MRSA samples from Consorcio Hospital General Universitario de Valencia (CHGUV) resistant to each of the antibiotics tested. MRSA, methicillin-resistant Staphylococcus aureus.
Characterization by MLST, spa typing, and SCCmec
We obtained complete MLST allelic profiles for the 190 samples (Supplementary Table S4). In total, 47 different STs were identified: 164 samples were assigned to 22 existing STs and 26 samples to 25 new STs that include 4 new alleles (Supplementary Table S5). The most abundant type in the hospital was ST125, with 46 cases (24.2%), followed by ST2628 (28 cases, 14.7%), ST8 (27 cases, 14.2%), ST5 (20 cases, 10.5%), and ST22 (16 cases, 8.4%). The rest of the STs were detected in 5 or fewer isolates.
The distribution of STs changed through time. While ST8, ST125, ST5, and ST2628 were detected throughout the analyzed period (2013–2018), we observed the appearance and disappearance of certain STs, such as ST22 (disappeared in 2016, but detected again in 2017), ST6187 (only detected in 2016), or ST1181 (emerging in the hospital between 2015 and 2016) (Supplementary Fig. S1).
Regarding the spa characterization, we obtained the spa type for 164 (86%) of the samples. The most abundant spa type in this study was t067 (N = 38), followed by t002 (N = 32), t008 (N = 24), and t022 (N = 16). In addition, 37 other spa types were identified, each of them present in fewer than 5 isolates. The spa typing showed a high diversity for this gene in several of the major STs of the hospital (Table 1). Seven STs were related to more than one spa type, especially those most represented, such as ST125 (10 spa types), ST5 (10 spa types), or ST2628 (5 spa types).
Most Prevalent Clones of Methicillin-Resistant Staphylococcus aureus Identified in the Study Dataset Considering Those STs and spa Types Identified in More Than 10 Isolates
New STs (new allelic combinations and/or new alleles) are not included.
In turn, some spa types have been detected in isolates with different STs. One of them is t067, found mainly not only in ST125 (CC5) but also in two isolates with ST5 (CC5) and ST1181 (CC8). The same occurred with spa types t002 (ST2628, ST125, and ST146) and t008 (ST8, ST1014, and ST1181).
The SCCmec type was obtained for 163 (85%) isolates. The SCCmec analysis revealed that 93.3% (152) of the isolates carried the SCCmec type IV, while only 11 isolates carried a different type (Table 1). SCCmec type II was present in five isolates belonging to ST5, ST125, ST1870, and ST7370. Type III was found in only two isolates belonging to ST72 and ST7372. Finally, type V was identified in four isolates: ST398 (two isolates), ST97, and ST7373.
Clonal distribution and antimicrobial susceptibility profiles
Each typing scheme on its own did not provide a high level of discrimination. However, the combination of the three schemes allowed the identification of 74 different clones. ST125-t067-IV was the most abundant clone in the study (27 cases) and it was detected at the hospital throughout the study period. The next most abundant clones were identified from 2014 onward and were ST2628-t002-IV, ST8-t008-IV, and ST22-t022-IV with 19, 16, and 16 cases, respectively. All these clones contain both HA-MRSA and CA-MRSA isolates. It is remarkable the identification of three isolates considered CA-MRSA that belong to clones related to livestock-associated MRSA (LA-MRSA) (ST398-t011-V and ST97-t267-V). Some of the STs have a large diversity of spa types and even different SCCmec types. Only ST22 (n = 16) is exclusively associated with one spa type (t022).
No association between clones and antimicrobial sensibility patterns was found (Fig. 2). Antimicrobial patterns were uniform between clones. Only isolates with spa type t008 had a different pattern, showing a higher number of isolates than the other clones resistant to gentamicin (74% vs. 18%, p < 0.001), mupirocin (74% vs. 21%, p < 0.001), amikacin (82.6% vs. 51.5%, p < 0.001), and tobramycin (82.5% vs. 52%, p < 0.001), and fewer resistant isolates to clindamycin (4.3% vs. 31.7%, p < 0.001) and erythromycin (17.4% vs. 45.5%, p < 0.001).
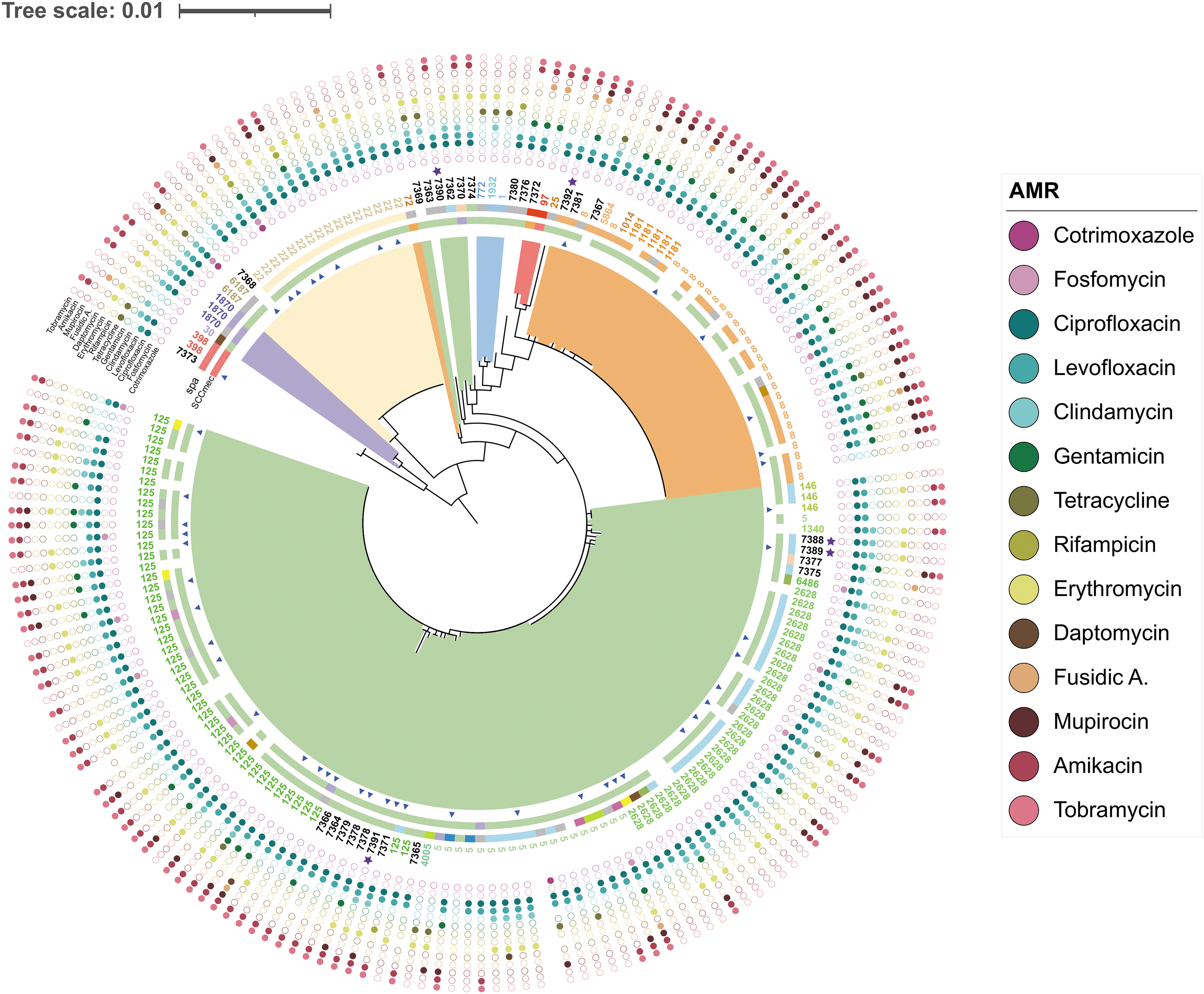
Maximum likelihood phylogenetic tree of 190 MRSA isolates from CHGUV obtained in the period 2013–2018. The color of the range indicates the CC to which each clade belongs. Blue triangles indicate isolates identified as HA-MRSA. The outer bands represent (from the inside to the outside) the SCCmec typing and the spa typing. Labels indicate the STs to which each isolate belongs. Outer blue stars indicate which isolates have new alleles. The outer dots indicate the antibiotics for which each isolate shows resistance, omitting those to which all or no isolates are resistant. CC, clonal complex; HA-MRSA, health care-associated MRSA.
Phylogeny and epidemiology
The phylogenetic tree for the 190 CHGUV isolates shows the relationships among the different MRSA clones in this hospital (Fig. 2). Most of the isolates belong to three clonal complexes: CC5, CC8, and CC22, in decreasing order of prevalence. These major clonal complexes comprise 91% of the isolates, 32 STs, and 32 spa types.
In CC5, the predominant STs were ST5, ST125, and ST2628 (49.5% of the total isolates). Moreover, 18 minority STs were identified in this CC. Similarly, the diversity of spa types is remarkable (23 spa types), although t067 (18.9% of the total isolates) predominated. CC5 is the only clonal complex with a paraphyletic distribution, being highly related to CC1 (three isolates with three STs and two spa types). Remarkably, most new STs (16 of 26 isolates) were found in CC5. In contrast, CC8 and CC22 had a monophyletic structure. They contained only a few STs (8 in CC8 and 3 in CC22) and spa types (8 and 3, respectively).
The least represented clonal complexes (CC1, CC30, and CC97) contained seven STs and 6 spa types. Only ST1870 was present in more than one isolate (three isolates) and was associated with spa types t012 and t018 and SCCmec types II and IV. No relationship was detected between the different clones and the sample type or hospital ward.
To gain a better understanding of the genetic variation at this hospital in the Spanish context, we obtained an ML tree using the concatenated gene sequences of MLST from our isolates and 257 additional Spanish S. aureus isolates. The phylogenetic tree (Supplementary Fig. S2) shows that most clonal complexes in the Valencian hospital have also been identified in other Spanish regions, especially in Castille-Leon and Catalonia. Methicillin-resistant isolates were most prevalent in the largest clonal complexes (CC5, CC22, and CC8).
Discussion
MRSA is one of the main causative agents of infections in health care settings and, in recent decades, it has increasingly been related to community transmission. 30 Some strains are restricted to a geographical area, while others have a global spread. The reduction of infection rates depends on the prevention of its transmission, for which the detection of the entry of new strains and their containment is essential. 14 Molecular epidemiology techniques provide an appropriate method for this end. 31
In this work, we have described the molecular epidemiology of MRSA isolates obtained at the CHGUV from 2013 to 2018. These isolates were characterized using three different molecular typing tools: MLST, spa typing, and SCCmec typing, to study the population and dynamics of the pathogen in the hospital. The combination of the three typing schemes provides a precise characterization of circulating clones. 31
In Spanish hospitals, the most prevalent MRSA clonal complexes are CC5, CC8, CC22, and CC30, although their frequencies have fluctuated over time. 32 In the early 2000s, ST125 (CC5) was responsible for more than 50% of infections caused by nosocomial MRSA strains in this country, ST125-SCCmec IV being one of the most prevalent clones, with a strong correlation with spa type t067. 33 This clone emerged in Spain in 1996 and soon became the most prevalent in Spanish hospitals, replacing ST247-SCCmec I, the Iberian clone. 34
Despite the high heterogeneity of STs identified in this study, around 90% of isolates belong to the aforementioned three major clonal complexes: CC5 (61% of our isolates), CC8 (20%), and CC22 (10%). 32 Most isolates in CC5 possess SCCmec IV, ST125-SCCmec IV being the most common clone mainly associated with spa type t067. This spa type is present in the CHGUV in both CA-MRSA and HA-MRSA throughout the study period (2013–2018), which suggests a possible transmission from the community.
Many of the remaining isolates also possess SCCmec IV, and most of them correspond to CC5, CC22, and CC8. ST5-SCCmec IV (the so-called “Pediatric Clone”) and ST22-SCCmec IV are two of the most prevalent and widely distributed HA-MRSA clones internationally. 32 The molecular type ST22-SCCmec IV corresponds to a pandemic strain of CC22, which causes most nosocomial infections in United Kingdom, but is also related to community transmission and zoonoses. 3 This molecular type represents 8.4% of isolates from the Valencian hospital. We detected this clone in 2014 producing hospital-acquired infections. However, in later years (2015, 2017, and 2018), it was only identified in CA-MRSA, suggesting possible transmission between the hospital and the community.
The molecular type ST8-SCCmec IV, internationally known as USA300, predominantly presents community transmission, but nosocomial transmission has also been reported. 3 It is rare in Europe, but has previously been found in Switzerland and Spain.3,34 This ST is found in 10% of the CHGUV isolates, five of them HA-MRSA, and shows a strong correlation with the spa type t008, as previously determined.3,34
In addition, the presence of two CA-MRSA STs (ST398 and ST97) at the CHGUV correlated with zoonotic variants of MRSA is remarkable. On the one hand, the two ST398 isolates belong to the clone ST398-t011-SCCmec V, which is related to resistance to tetracycline, an antibiotic widely used in livestock. 35 This clone has been identified in several countries, including Italy, Germany, the United States, and Spain. 3 On the other hand, the only ST97 isolate corresponds to the clone ST97-t267-SCCmec. V. Harrison et al 35 showed that two ST97 isolates obtained in United Kingdom came from a human lineage that arose from a zoonotic event 40 years ago, so they had no direct association with animal sources. Further in-depth analysis of these isolates should be performed to study their possible association with cattle.
Antimicrobial susceptibility patterns were mostly uniform among isolates. Isolates showed low sensibility patterns to several antimicrobial categories, including those prescribed for the treatment of severe MRSA infections or as secondary-line treatment when vancomycin fails. 36 The worldwide increase of clinical MRSA isolates with reduced susceptibility to vancomycin 37 leads to a greater need for alternative antibiotics as treatment. In our study, we detected six CA-MRSA isolates resistant to daptomycin belonging to four different STs (ST8, ST1181, ST5, and ST125). This finding supports the need to reinforce control measures and surveillance strategies in the HGUV.
Although we have provided an accurate description of the MRSA population in the hospital over 6 years, to verify the possible reintroduction in the hospital of persistent strains from the community, it would also be advisable to establish a surveillance system integrating genomic and epidemiological data. 38
Conclusions
The molecular characterization of MRSA isolates performed in this study has made it possible to identify the prevalent clones in CHGUV. In addition, it allowed us to detect the continuous emergence of low-incidence strains. The high proportion of resistant MRSA clones detected and the presence of LA-MRSA variants of zoonotic origin emphasize the need for surveillance. As demonstrated in this study, epidemiological and molecular surveillance provide a powerful tool to detect and track the emergence and spread of clinically relevant MRSA clones.
Footnotes
Authors' Contributions
A.S.S.: writing – original draft; data curation; formal analysis; and visualization; N.G.G.: conceptualization; methodology; formal analysis; writing—review and editing; supervision; and visualization; D.B.: data curation; P.R.H.: data curation; R.V.: data curation; I.C.B.: data curation; N.T.: resources; C.S.: resources; C.G.: conceptualization and resources; F.G.C.: conceptualization; funding acquisition; methodology; writing—review and editing, and supervision.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by project BFU2017-89594R from MICIN (Spanish Government) and Conselleria de Sanitat Universal i Salut Publica (Generalitat Valenciana).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.