
Abstract
Artificial RNA reagents such as small interfering RNAs (siRNAs) and aptamers often must be chemically modified for optimal effectiveness in environments that include ribonucleases. Mycoplasmas are common bacterial contaminants of mammalian cell cultures that are known to produce ribonucleases. Here we describe the rapid degradation of nuclease-stabilized RNA oligonucleotides in a human embryonic kidney 293 (HEK) cell culture contaminated with Mycoplasma fermentans, a common species of mycoplasma. RNA with 2′-fluoro- or 2′-O-methyl- modified pyrimidines was readily degraded in conditioned media from this culture, but was stable in conditioned media from uncontaminated HEK cells. RNA completely modified with 2′-O-methyls was not degraded in the mycoplasma-contaminated media. RNA zymogram analysis of conditioned culture media and material centrifuged from the media revealed several distinct protein bands (ranging from 30 to 68 kDa) capable of degrading RNA with 2′-fluoro- or 2′-O-methyl-modified pyrimidines. Finally, the mycoplasma-associated nuclease was detected in material centrifuged from the contaminated culture supernatants in as little as 15 minutes with an RNA oligo-containing 2′-O-methyl-modified pyrimidines and labeled with a 5′-fluorescein amidite (FAM) and 3′-quencher. These results suggest that mycoplasma contamination may be a critical confounding variable for cell culture experiments involving RNA-based reagents, with particular relevance for applications involving naked RNA (e.g., aptamer–siRNA chimeras).
Introduction
These modifications are widely employed in the development of small interfering RNAs (siRNAs) and RNA aptamers for both research and therapeutic applications (BEHLKE, 2008; Thiel and Giangrande, 2009). siRNAs can be modified with 2′-O-methyl substitutions in both sense and antisense strands without loss of silencing potency, but only a subset of nucleotides are typically modified with 2′-O-methyls, as over-modification of the siRNA can reduce or eliminate its silencing ability (Chiu and Rana, 2003; Czauderna et al., 2003; Harborth et al., 2003; Choung et al., 2006; Santel et al., 2006a; Santel et al., 2006b). siRNAs with 2′-fluoro modified pyrimidines have also been reported to retain silencing activity in vitro as well as in vivo (Layzer et al., 2004; Allerson et al., 2005; Prakash et al., 2005; Kraynack and Baker, 2006).
RNA with 2′-fluoro modified pyrimidines is the most commonly used chemistry for development of RNA aptamers with potential therapeutic applications. Such RNA is stable in animal serum and can also be efficiently transcribed in vitro with a mutant viral RNA polymerase, thus facilitating its use in the aptamer discovery process known as SELEX (Systematic Evolution of Ligands by EXponential enrichment) (Ellington and Szostak, 1990; Tuerk and Gold, 1990; Pieken et al., 1991; Huang et al., 1997). The stability of this RNA in other contexts, such as conditioned cell culture media, has not been well studied.
Mycoplasmas are a genus of small bacteria that are common contaminants of cell cultures (Drexler and Uphoff, 2002). They lack a cell wall, are not susceptible to the antibiotics usually employed in cell culture, and often go undetected in cell culture due to their small size (Drexler and Uphoff, 2002). Some mycoplasma species, notably Mycoplasma pneumoniae, are human pathogens (CUNHA, 2010). Various mycoplasma species are known to produce ribonucleases and deoxyribonucleases (e.g., Minion and Goguen, 1986; Marcus and Yoshida, 1990; Minion et al., 1993; el-Farrash et al., 1994; Shang et al., 1995; Bendjennat et al., 1997); however, the ability of these nucleases to degrade chemically modified RNA has not been explored.
In the course of developing various cell-based assays for RNA aptamers, we evaluated the stability of RNA with 2′-fluoro modified pyrimidines in cell culture media conditioned by human embryonic kidney 293 (HEK) cells and found the RNA to be substantially degraded after fairly brief incubations. We subsequently realized that the HEK cells were contaminated with mycoplasma. We then sought to determine whether this contamination was responsible for the nuclease activity.
Materials and Methods
Cell culture and conditioned media
HEK cells were inadvertently contaminated with mycoplasma at some point during routine culture maintenance. The contamination was later detected and confirmed by polymerase chain reaction (PCR) to be M. fermentans. This mycoplasma contaminated cell line was used as a positive control for mycoplasma testing methods. Uncontaminated HEK cells were obtained from ATCC (ATCC-CRL-1573™). Contaminated and uncontaminated HEK cells were grown in Dulbecco's Modified Eagle Medium (DMEM; GIBCO) containing 10% heat-inactivated bovine serum, 50 U/mL penicillin, and 50 μg/mL streptomycin at 37°C, 5% CO2 in a moist atmosphere. For preparation of conditioned media, contaminated or uncontaminated HEK cells were grown to ∼80% confluency on 100-mm or 150-mm culture dishes. The culture media was then replaced with fresh media. After 48 hours of incubation, the media was centrifuged for 6 minutes at 1,250 rpm in a tabletop centrifuge to remove cellular debris. Finally, the supernatant was transferred into a fresh tube and used as conditioned media. Unconditioned media had the same composition (see above), but was not incubated with cells. To collect particulate material from the media, six milliliters of conditioned media was centrifuged at 13,300 rpm in a microcentrifuge. The pellet was washed with phosphate buffered saline (PBS) and then dissolved in 20 μL of 1% Triton X-100 in PBS.
Mycoplasma culture
Mycoplasma broth consisted of 10% yeast extract solution (Gibco), 20% heat-inactivated fetal bovine serum, 70% heart infusion broth (BD Biosciences), 50 U/mL penicillin, and 50 μg/mL streptomycin. A freeze-dried culture of M. fermentans (ATCC# 15474) was rehydrated in 10 mL of mycoplasma broth. Several 10-fold serial dilutions of this culture were then prepared and the bacteria were grown in 50-mL conical tubes at 37°C for several days. For bacterial lysate preparation, 1 mL of M. fermentans culture grown for 5 days was pelleted at 13,300 rpm for 5 minutes. Supernatant was discarded and the lysate was prepared by dissolving the pellet in 20 μL of 1% Triton X-100 in PBS. The lysate was incubated with RNase substrates for 1 hour at 37°C.
Chemically modified RNA
The following 51-nucleotide-long RNA sequence was used for the gel-based degradation assays and the RNA zymograms: 5′-GGGAGGACGAUGCGGGACUAGCGAUCUGUUACGCACAGACGACUCGCCCGA-3′. Several versions of this RNA, with modifications as described in figure legends and the results section, were used. Fluorescein amidite (FAM)-labeled versions were used in the gel-based degradation assays, whereas nonfluorescent versions were used for the zymograms. These RNAs were obtained from Trilink Biotechnologies.
Gel-based degradation assay
For each degradation assay sample, 6 μL of oligo (2 μM) was combined with 6 μL of unconditioned and conditioned media and incubated for 0.5, 1, 2, or 4 hours at 37°C. After incubation, samples were combined with 12 μL of loading buffer (formamide with 0.5×TBE), incubated for 6 minutes at 65°C, transferred to ice for 5 minutes, briefly centrifuged, and kept on ice until loading. Samples were run on a 7.7-M urea, 10% acrylamide gel at 100 volts for 80 minutes. Gel images were acquired with a Gel Doc™ XR+ System (Bio-Rad) with ultraviolet light transillumination and a standard fluorescence filter for imaging ethidium bromide.
RNase substrate plate-reader assays
The RNase substrates were synthesized by Integrated DNA Technologies (IDT). These probes consist of a 12-nucleotide-long RNA oligo, 5′-UCUCGUACGUUC-3′, with the chemical modifications indicated in figure legends, flanked by a FAM (5′ modification) and a pair of fluorescence quenchers, ZEN and Iowa Black (3′ modifications). For the RNA degradation assays, 1 μL of each RNase substrate (50 picomoles) was combined with 9 μL of sample (e.g., conditioned media) and incubated at 37°C for time points indicated in the figures. After the incubation period, 290 μL of PBS supplemented with 10 mM EDTA and 10 mM EGTA was added to each sample, and 95 μL of each sample was loaded in triplicate into a 96-well plate (96F non-treated black microwell plate). Fluorescence intensity was measured with a fluorescence microplate reader (Analyst HT; Biosystems). To visualize the Triton X-100 lysate samples, the undiluted 10 μL reactions were imaged in eppendorf tubes with a Gel Doc™ XR+ System (Bio-Rad) with ultraviolet light transillumination and a standard fluorescence filter for imaging ethidium bromide.
PCR
All cell cultures were tested for mycoplasma infection by PCR. Four primer sets, previously described (Choppa et al., 1998), were used to identify a conserved region among all members of the genus mycoplasma and 3 specific species: M. fermentans, Mycoplasma hominis, and Mycoplasma penetrans. Mycoplasma, centrifuged from conditioned cell culture media, provided the template DNA for the mycoplasma-specific PCRs. The mycoplasma was isolated from the culture media as follows: conditioned media was centrifuged for 6 minutes at 1,250 rpm to pellet cellular debris. Two milliliters of the supernatant from this spin were then centrifuged for 5 minutes at 17,000 g to pellet any mycoplasma present in the media. The pellet was resuspended in 50 μL of water; this served as the PCR template. The PCR reaction mixtures were prepared in a total volume of 100 μL containing 10 μL of PCR template, 2 μL dNTPs (10 μM), 1 μL of each primer (100 μM), 50 μL of Choice Taq Mastermix (Denville Scientific) and 36 μL of water. Thirty cycles of PCR were carried out. The temperature steps were as follows: 94°C for 5 minutes, 30× (denaturation at 94°C for 30 seconds; annealing at 55°C for 30 seconds; elongation at 72°C for 30 seconds), and a final extension step of 72°C for 10 minutes was carried out at the completion of the cycling. The PCR products were analyzed on a 2% (w/v) agarose gel stained with 0.5 μg/mL ethidium bromide. The DNA bands were visualized with a Gel Doc™ XR+ System (Bio-Rad) with ultraviolet light transillumination and a standard fluorescence filter for imaging ethidium bromide.
DAPI staining
Mycoplasma was visualized in cultured cells via DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride; Invitrogen) staining. Briefly, HEK cells were grown on glass-bottomed culture dishes from MatTek. Cells were then fixed by incubation for 20 minutes at −20°C in 100% methanol and stained with DAPI following the protocol provided in the DAPI product insert. The cells were then rinsed several times in PBS and fluorescence was visualized with an Olympus IX71 fluorescence microscope equipped with a 40× oil-immersion objective (fluorescence filters appropriate for DAPI) and a cooled CCD digital camera.
DNA zymograms
Mycoplasma-free and mycoplasma-positive cell cultures were serum starved for 48 hours on 150-mm culture dishes in 20 mL of DMEM. Media was collected, spun at 1,250 rpm for 6 minutes to eliminate cell debris, and then spun at 17,000 g for 5 minutes to pellet mycoplasma from the conditioned media. These pellets were solubilized in 50 μL of sodium dodecyl sulfate (SDS) sample loading buffer. The supernatants from the second centrifugation were concentrated with a YM-10 Amicon centrifugal filter device (MW cutoff of 10 kDa) to a final volume of approximately 100 μL. One or three microliters of the concentrated media or pellet, respectively, were loaded per lane of an 8% acrylamide SDS gel containing 200 μg/mL salmon sperm DNA (Invitrogen). After the gel was run, nucleases were activated by a series of 10-minute washes: 2 washes in 2 mM CaCl2, 2 mM MgCl2, 2.5% Triton X-100 in water; 2 washes in 2 mM CaCl2, 2 mM MgCl2, 2.5% Triton X-100 in 50 mM Tris-HCl (pH 7.4); and 2 washes in 2 mM CaCl2, 2 mM MgCl2 in 50 mM Tris-HCl (pH 7.4). The gels were then incubated in 2 mM CaCl2, 2 mM MgCl2 in 50 mM Tris-HCl (pH 7.4) for either 2 hours or overnight at 37°C. The gels were stained with 0.5 μg/mL ethidium bromide and visualized with a Gel Doc™ XR+ System (Bio-Rad) with ultraviolet light transillumination and a standard fluorescence filter for imaging ethidium bromide.
Chemically modified RNA zymograms
Samples were prepared as for the DNA zymograms (described above). Proteins were separated on 8% acrylamide SDS gels polymerized with either 550 nM RNA oligo with 2′-fluoro-modified pyrimidines or 250 nM RNA oligo with 2′-O-methyl-modified pyrimidines. See the section Chemically Modified RNA for the sequence of these oligos. Gels were washed as above, but stained with a 1 per 10,000 dilution of SYBR® Gold Nucleic Acid Gel Stain (Invitrogen) and visualized with a Gel Doc™ XR+ System (Bio-Rad) with ultraviolet light transillumination and a standard fluorescence filter for imaging ethidium bromide.
Results
To evaluate the stability of RNA with 2′-fluoro modified pyrimidines in conditioned culture media, a 51-nucleotide RNA with 2′-fluoro modified pyrimidines and a 3′-FAM was incubated in serum-containing media, conditioned with an HEK cell culture recently obtained from ATCC, or with an older HEK cell culture (Fig. 1). As previously reported, this RNA was found to be resistant to nuclease degradation in the presence of animal serum (unconditioned media incubation; lane 1 in Fig. 1). While the modified RNA was also stable in conditioned media from the HEK cells obtained from ATCC, the conditioned media from the older HEK cells almost completely degraded it after a 4-hour incubation (lane 2, Fig. 1). A PCR assay for the presence of mycoplasma detected a DNA sequence conserved within the genome of the mycoplasma genus in the culture supernatant of the older HEK cell culture supernatant, but not in the media from the more recently obtained culture (Fig. 2). A DNA sequence specific to the M. fermentans species was also detected in media from the older HEK cell culture, as indicated with a PCR product of expected size (lane 3, Fig. 2). Sequencing of this PCR product confirmed that the amplified sequence is derived from the M. fermentans genome (not shown). In contrast, neither M. hominis nor M. penetrans was detected. As an additional means of detecting mycoplasma, DAPI staining of HEK cells from these 2 cultures was carried out (Fig. 3). Small, punctate, extra-nuclear DAPI labeling, indicative of mycoplasma contamination, was seen throughout the older HEK culture (Fig. 3A), but only nuclear labeling was seen in the culture obtained from ATCC (Fig. 3B). Together, these data demonstrate the presence of a ribonuclease that readily degrades RNA with 2′-fluoro modified pyrimidines in a mycoplasma-contaminated cell culture, but not in a mycoplasma-free HEK culture.
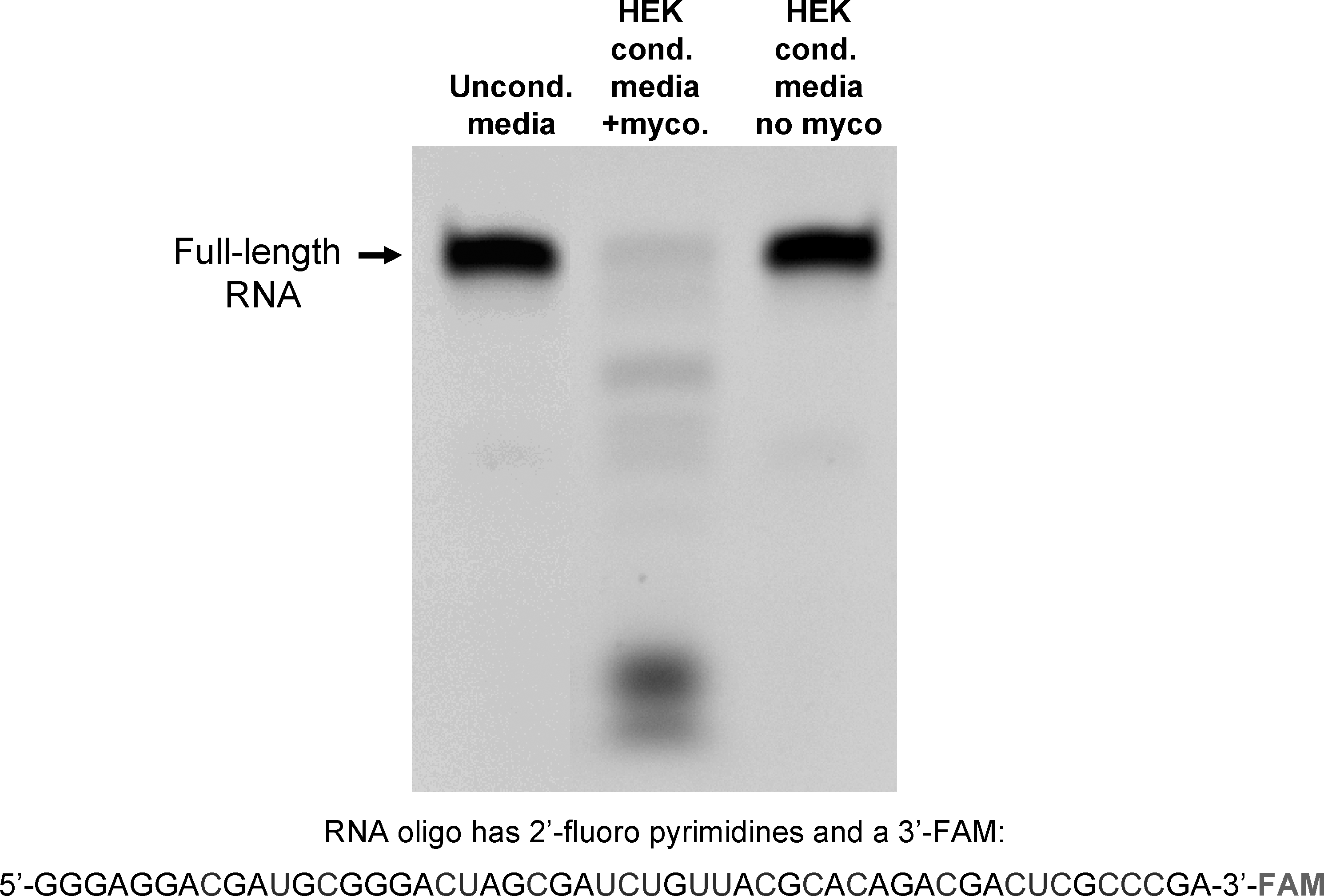
An RNA oligo with 2′-fluoro-modified pyrimidines is degraded in conditioned media from mycoplasma contaminated HEK cells. A 51 nucleotide-long RNA oligo, composed of 2′-fluoro modified pyrimidines and unmodified purines was incubated with unconditioned culture media, culture media conditioned by mycoplasma contaminated HEK cells, or culture media from uncontaminated HEK cells for 4 hours at 37°C. The RNA was then resolved on a urea-acrylamide denaturing gel and imaged with a digital camera and UV light transillumination; the oligo is labeled on its 3′ end with FAM.

Polymerase chain reaction (PCR)-based assay detected mycoplasma contamination in HEK cell culture and identified species as M. fermentans. PCR primer sets specific for genomic components of M. fermentans, Mycoplasma hominis, Mycoplasma penetrans, or for a region of the mycoplasma genome that is conserved within the genus were used to amplify DNA present in conditioned culture media. Expected PCR product sizes are as follows: mycoplasma genus PCR: 280 bp; M. fermentans: 206 bp; M. hominis: 170 bp; M. penetrans: 470 bp. Material centrifuged from one HEK cell culture was used as template for PCRs run on the gel on the left, while material centrifuged from a second HEK cell culture recently obtained from ATCC was used as template for PCRs run on the gel on the right.
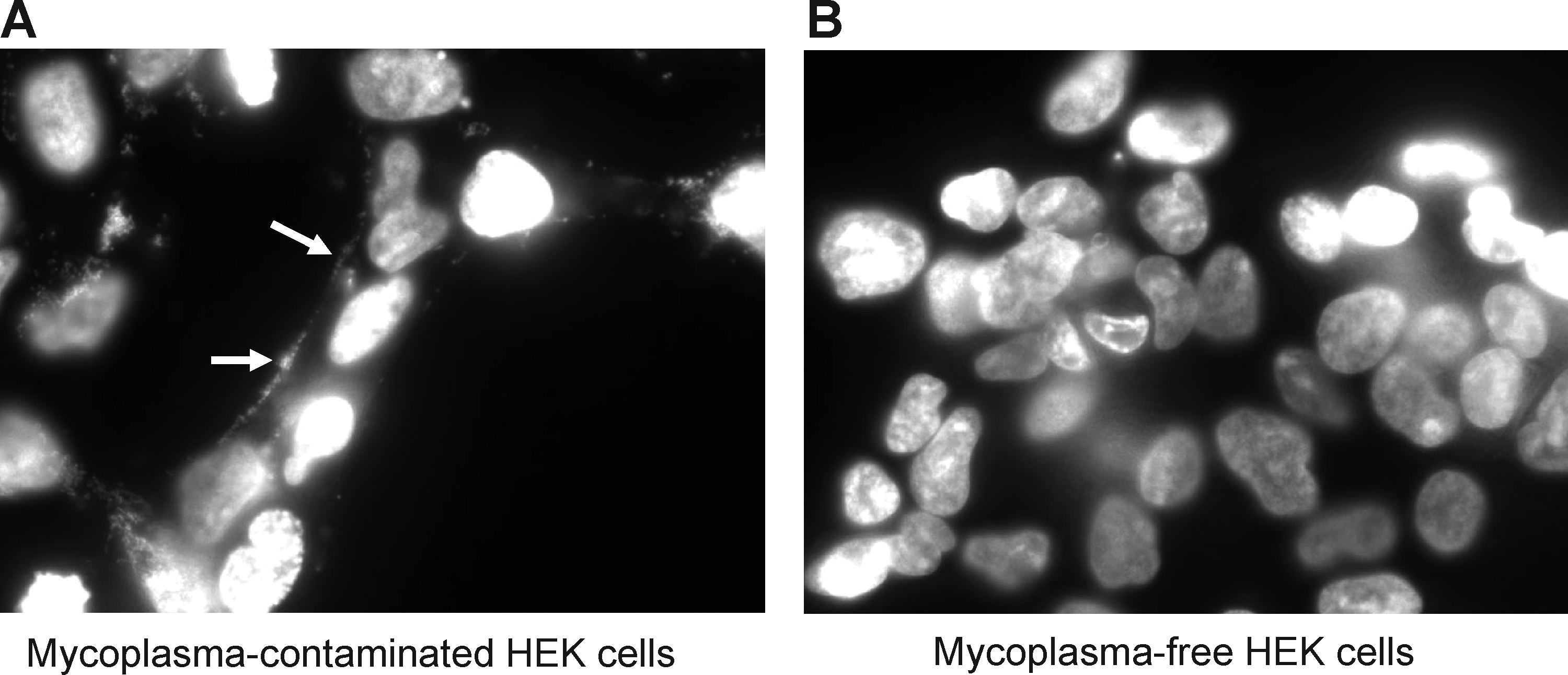
Mycoplasma contamination detected in HEK cells with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) stain. HEK cells from 2 cultures—one older culture
Next we studied the activity of this ribonuclease after heat pretreatment, in the presence of EDTA (a chelator of divalent cations), and in the presence of Superase.in (a broad-spectrum ribonuclease inhibitor) (Fig. 4). The same RNA oligo initially examined (Fig. 1) was used for this experiment (51-mer with 2′-fluoro-modified pyrimidines and a 3′-FAM), with conditioned media from the mycoplasma-contaminated HEK cells. Results of this experiment indicate that the ribonuclease activity is sensitive to heat treatment (lane 3 in Fig. 4) and is thus likely protein in nature. Like many ribonucleases, its activity is dependent on divalent cations, as chelation of divalent cations with EDTA inhibited degradation of the modified RNA (lane 4, Fig. 4). However, the broad-spectrum RNase inhibitor, Superase.in, did not have an apparent impact on the activity (lane 5, Fig. 4).
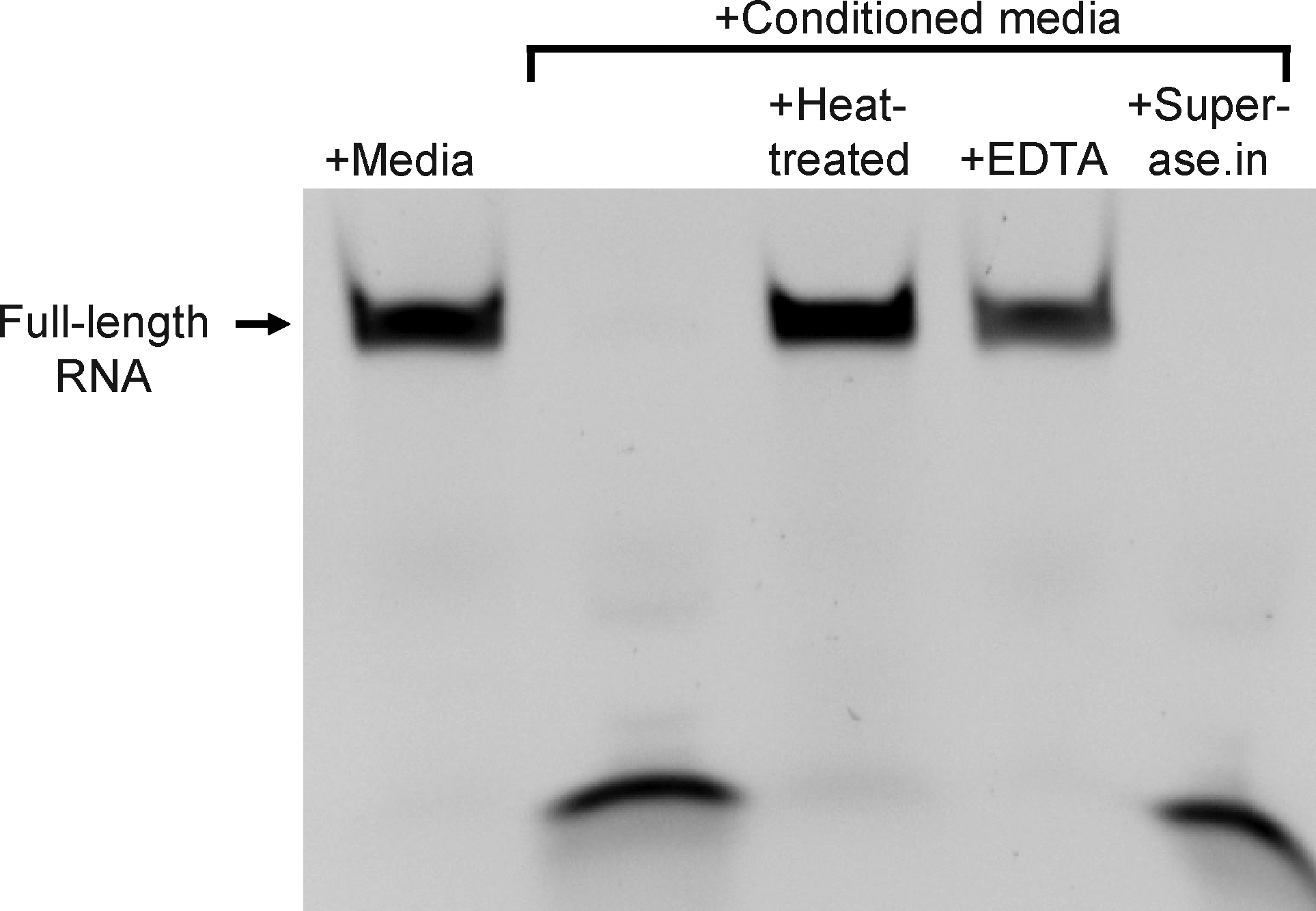
Nuclease activity in mycoplasma-contaminated culture media is sensitive to heat treatment and depends on the presence of divalent cations. A 51-nucleotide-long RNA oligo, composed of 2′-fluoro modified pyrimidines and unmodified purines was incubated for 4 hours at 37°C, either in fresh culture media or culture media conditioned by mycoplasma contaminated HEK cells. To determine sensitivity of the nuclease activity to heat, conditioned media was heated to 95°C for 10 minutes prior to combination and incubation with RNA. To determine the dependence of the nuclease activity on divalent cations, conditioned media was incubated with RNA in the presence of 10 mM EDTA. A broad-spectrum RNase inhbitor, Superase.in, was coincubated (at 1 U/μL) with the RNA in conditioned media to determine the sensitivity of the nuclease activity to this reagent. The RNA was resolved on a urea-acrylamide denaturing gel and imaged with a digital camera and UV light transillumination; the oligo is labeled on its 3′ end with FAM.
While the 2′-fluoro nucleotide modification is widely used for the development of RNA aptamer-based therapeutic approaches, other modifications are more commonly used to protect synthetic RNAs in other applications such as RNA interference (RNAi). The 2′-O-methyl modification is widely employed, and thus we examined the susceptibility of RNA with 2′-O-methyl-modified nucleotides to degradation by the mycoplasma-associated ribonuclease activity. For this experiment, 3 RNA oligos were used. Each was 51 nucleotides in length, of identical sequence, and with a 3′-FAM. The first oligo had 2′-fluoro-modified pyrimidines (purines were unmodified; described above), the second had 2′-O-methyl-modified pyrimidines (purines were unmodified), and every nucleotide of the third oligo was modified with 2′-O-methyls. Coincubation of these oligos with media conditioned by the mycoplasma-contaminated HEK cells for 30 minutes, 1 hour, 2 hours, or 4 hours again demonstrated the near-complete degradation of the oligo with 2′-fluoro-modified pyrimidines (lanes 1 and 2 in Fig. 5A–D). The oligo with 2′-O-methyl-modified pyrimidines was more resistant to degradation, but was almost completely degraded after 4 hours (lanes 3 and 4 in Fig. 5A–D). Finally, there was no detectable degradation of the oligo that was completely modified with 2′-O-methyls at any of the time-points (lanes 5 and 6, Fig. 5A–D).
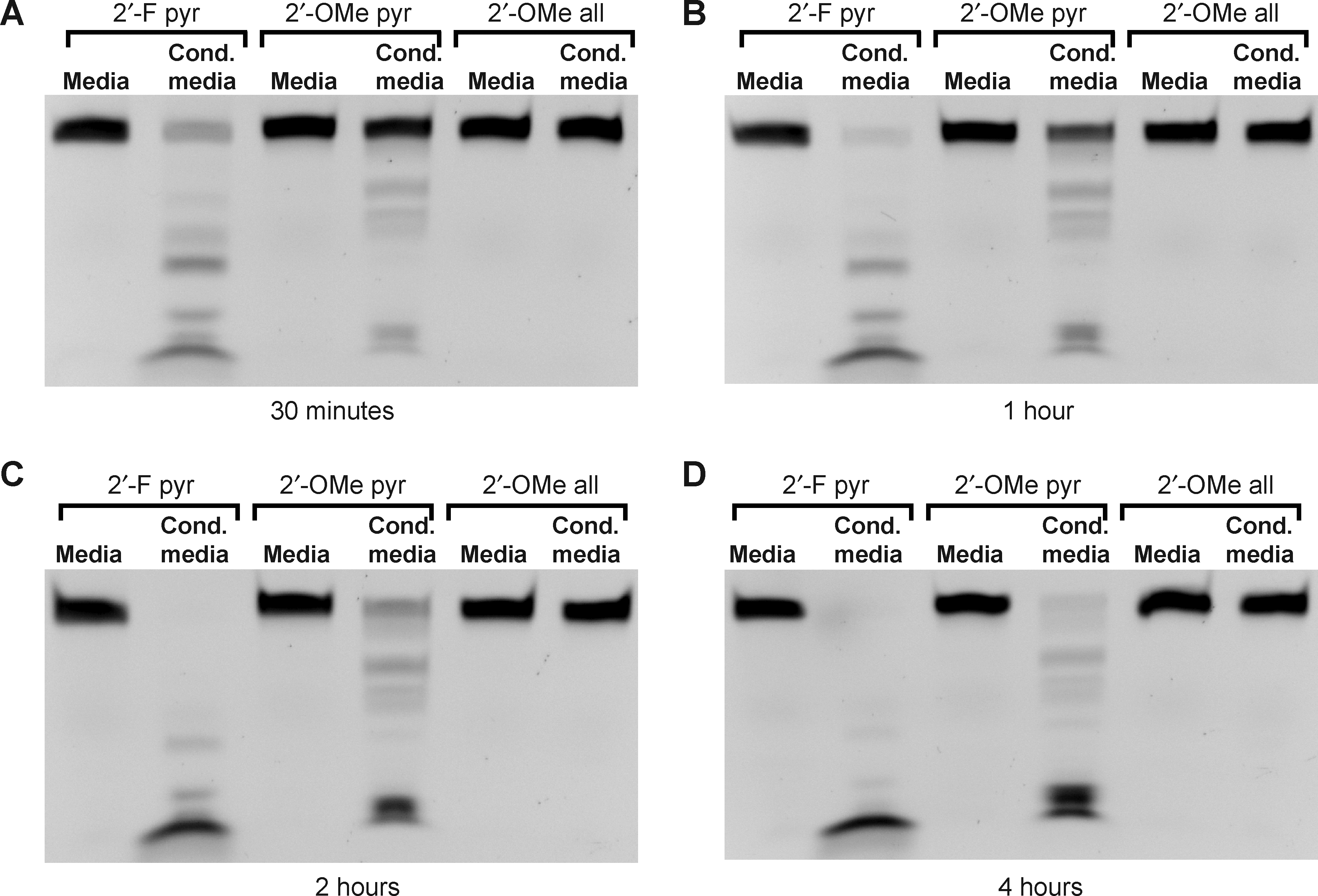
Time-course and substrate specificity of nuclease activity in mycoplasma contaminated culture media. Three 51-nucleotide RNA oligos of identical sequence but with different chemical modifications (2′-fluoro modified pyrimidines only, 2′-O-methyl modified pyrimidines only, or complete 2′-O-methyl modification) were incubated with fresh culture media or with culture media conditioned by mycoplasma contaminated HEK cells at 37°C for 30 minutes
Additional cell lines, contaminated with distinct mycoplasma species (i.e., non-M. fermentans species) were also tested for nuclease activity against RNA oligos with 2′-O-methyl- and 2′-fluoro- modified pyrimidines. Some of these cell lines possessed strong nuclease activity in their supernatants while others did not (not shown).
To further characterize the mycoplasma-associated ribonuclease, we carried out zymograms with unmodified DNA or RNA with 2′-fluoro- or 2′-O-methyl- modified pyrimidines (Fig. 6). For these experiments, we used serum-free conditioned cell culture media that was concentrated with filter centrifugation, as well as detergent-lysed particulate matter centrifuged from the conditioned media. The particulate matter presumably contains mycoplasma in the contaminated culture sample and was also found to possess ribonuclease activity.

Nuclease sizes determined with zymograms. Concentrated media or material pelleted by centrifugation from media conditioned by mycoplasma-free or mycoplasma-contaminated HEK cells was resolved on 8% acrylamide-sodium dodecyl sulfate (SDS) gels embedded with DNA
Multiple protein bands present in the mycoplasma-contaminated but not the mycoplasma-free concentrated culture supernatants could be seen in all 3 zymograms (lanes 2 and 3 in Fig. 6A–C). A cluster of bands that migrated between the 37- and 50-kDa molecular weight markers was prominent in these samples. A smaller protein, of approximately 30 kDa, digested the modified RNAs; no digestion was seen in this region of the DNA zymogram. A larger protein, of approximately 68 kDa, produced a band in the DNA zymogram, but not in the modified RNA zymograms. However, longer digestion periods (e.g., overnight) did yield a band of approximately this size in the modified RNA zymograms (not shown). The prominent cluster of bands between 37 kDa and 50 kDa present in all of the zymograms suggests the presence of multiple nucleases with broad substrate specificities.
The detergent-lysed particulate matter produced a very different pattern of bands on the zymograms (lanes 4 and 5, Fig. 6A–C). As with the concentrated supernatant, dark bands indicating digestion were only seen in the sample prepared from the mycoplasma-contaminated culture; however, the patterns of bands clearly indicate that there are multiple nucleases present in the particulate matter of the culture media with distinct substrate specificities. The prominent cluster of bands between the 37-kDa and 50-kDa molecular weight markers that was present in the concentrated culture supernatant was not seen in the particulate matter samples.
Because the mycoplasma-associated nuclease can be distinguished from endogenous mammalian nucleases by its distinct substrate specificity, we reasoned that the presence of mycoplasmas, in various contexts, might be inferred by the susceptibility of chemically modified ribonuclease substrates to degradation. While gel-based assays provide a simple and straightforward means of detecting nuclease activity, a more rapid and sensitive assay for ribonucleases that degrade unmodified RNA has been described. The basis for this assay is a short oligonucleotide RNase substrate, end-labeled with a fluorophore on one end that is rendered nonfluorescent by its close proximity to a quencher on the other end (Fig. 7A) (Kelemen et al., 1999). Upon cleavage of the substrate, the quencher diffuses away from the fluorophore, which then exhibits fluorescence.

Rapid detection of mycoplasma-associated nuclease activity with chemically modified RNase substrates.
This approach was adapted to detect the mycoplasma-associated nuclease by generating chemically modified RNase substrates with fluorophore and quencher conjugates. Four different RNA chemistries were tested: 2′-fluoro-modified pyrimidines, 2′-O-methyl-modified pyrimidines, complete 2′-fluoro modifications, and complete 2′-O-methyl modifications. Initially, each of these RNase substrates was incubated for 4 hours with culture media conditioned by either mycoplasma-free or mycoplasma-contaminated HEK cells (Fig. 7B). While the complete 2′-O-methyl-modified substrate was not digested in either media, the other 3 RNase substrates exhibited substantially greater fluorescence after incubation in the mycoplasma-contaminated media. Of these, the substrate with 2′-O-methyl-modified pyrimidines exhibited the greatest relative fluorescence increase between uncontaminated and contaminated media. This substrate was thus characterized further.
Centrifugation of particulate matter from the mycoplasma-contaminated culture supernatants provided a simple and rapid means of obtaining concentrated nuclease activity. We explored the possibility that this approach might increase the sensitivity of mycoplasma detection with RNase substrates. The RNase substrate with 2′-O-methyl-modified pyrimidines was incubated with a detergent lysate of centrifuged particulate matter from mycoplasma-free or mycoplasma-contaminated HEK cells for 1 hour (Fig. 7C). Conditioned media from these cultures was compared in parallel. While both the lysate and conditioned media exhibited strong nuclease activity, the lysate produced a greater signal in this assay. A clear signal could be seen over the background in this assay, even with an abbreviated (15 minutes) incubation (Fig. 7D, fluorescence was measured on an ultraviolet light box in this case).
The contaminated HEK cell culture is a complex preparation, as it contains cells derived from two distinct organisms. While the uncontaminated HEK cells lack nuclease activity capable of efficiently degrading RNA with 2′-fluoro-modified or 2′-O-methyl-modified pyrimidines, we also sought to measure such activity in a pure culture of M. fermentans. Consistent with our observations of the contaminated HEK cell culture, lysates prepared from M. fermentans bacteria exhibited robust nuclease activity against RNA substrates with 2′-fluoro-modified and 2′-O-methyl-modified pyrimidines (Fig. 8). Nuclease activity against RNA substrates with 2′-fluoro-modified and 2′-O-methyl-modified pyrimidines was also found in the bacterial culture supernatant (not shown).
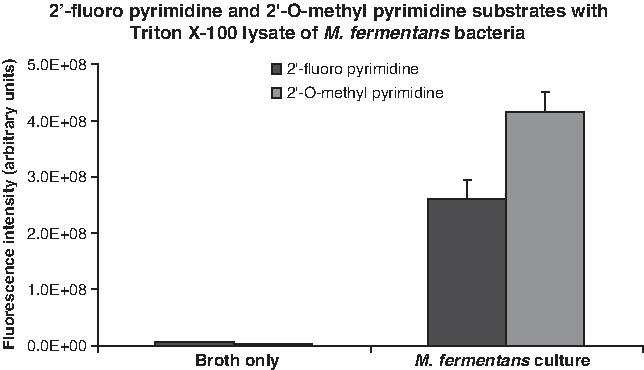
Nuclease activity in a Mycoplasma fermentans lysate. RNase substrates with the indicated compositions were incubated with a lysate of M. fermentans bacteria for 1 hour at 37°C, and fluorescence levels were measured with a fluorescence plate reader. A mock lysate prepared from centrifuged, bacteria-free broth served as negative control. Background fluorescence levels (determined by measuring the fluorescence level of each RNase substrate incubated in phosphate buffered saline have been subtracted from each experimental value.
Discussion
We found that conditioned media from HEK cells contaminated with M. fermentans possesses ribonuclease activity that readily degrades RNA with 2′-fluoro-modified pyrimidines and 2′-O-methyl-modified pyrimidines. Comparable ribonuclease activity was seen in a pure culture of M. fermentans, but not in an uncontaminated HEK cell culture. These observations are consistent with the conclusion that the activity in the contaminated HEK cell culture is derived from the mycoplasma bacteria. Zymograms with chemically modified RNA revealed the presence of multiple protein bands (from ∼30 kDa to ∼68 kDa) in the conditioned, mycoplasma-contaminated culture media that possess this ribonuclease activity. Some of these proteins were found in particulate matter in the media, which presumably contains free-floating mycoplasma. RNase substrates synthesized with chemically modified RNA detected the presence of this ribonuclease activity after a 15-minute incubation.
This work was undertaken to better understand the stability of chemically modified RNA oligonucleotides in cell culture settings. The results identify a potentially critical variable for the use of RNA-based reagents in cell culture experiments. While the potential for mycoplasma-based artifacts in cell culture experiments is widely known, many researchers do not regularly test their cell lines for mycoplasmas (Drexler and Uphoff, 2002). Lack of regular testing is perhaps the primary reason that mycoplasmas continue to be problematic for cell culture studies. It is estimated that 15–35% of cell lines used are contaminated with mycoplasmas, with M. fermentans among the most prevalent species identified in cell lines (Drexler and Uphoff, 2002).
The manner in which mycoplasma contamination affects experimental outcome will vary depending on the nature of the experiment. Our results suggest that experiments involving the application of naked RNA directly to cells would be particularly vulnerable to mycoplasma-dependent RNA degradation and experimental failure. These experiments include the study or application of siRNAs, RNA aptamers, and ribozymes. For instance, the delivery of siRNAs by directly coupling them to targeting reagents such as antibodies, aptamers, peptides and other synthetic ligands all entail the application of naked RNA to cells (Song et al., 2005; McNamara et al., 2006; Kumar et al., 2007; Zhou et al., 2008; Kortylewski et al., 2009; Pastor et al., 2010). The evaluation of RNA aptamers targeting cell surface receptors or extracellular targets such as growth factors in cell culture, likewise, involves the application of naked RNA to cells (Cerchia et al., 2005; Pestourie et al., 2005; McNamara et al., 2008).
The characterization of the mycoplasma-associated ribonuclease activity with nucleic acid zymograms revealed the presence of multiple proteins with deoxyribonuclease and ribonuclease activity in the contaminated cell culture media. The presence of multiple bands in similar patterns in all 3 of the zymograms between the 37-kDa and 50-kDa molecular weight markers suggests that several nucleases capable of digesting DNA, or RNA with 2′-fluoro- or 2′-O-methyl- modified pyrimidines, are produced by the mycoplasma. Other bands found in the mycoplasma-contaminated samples exhibited substrate -specific degradation among the 3 nucleic acid chemistries tested. Altogether, the results from the zymograms suggest that there are multiple mycoplasma-derived ribonucleases present in the contaminated culture media that can readily degrade RNA with either 2′-fluoro- or 2′-O-methyl- modified pyrimidines. The identity of these proteins has not been determined. Because zymograms depend on protein refolding following SDS denaturation, it is possible that some ribonucleases present in the culture media did not yield a signal on the zymograms.
The fraction of mycoplasma species that produce ribonucleases capable of degrading chemically modified RNAs is uncertain. The fact that RNA oligos with 2′-fluoro- or 2′-O-methyl- modified pyrimidines were degraded in cultures contaminated with distinct yet unidentified species of mycoplasmas indicates that the activity is not limited to the M. fermentans species. Considering the diverse nature of mycoplasmas, it is perhaps not surprising that we found mycoplasma-contaminated cell cultures that lacked such robust nuclease activity.
Because many mycoplasmas are human pathogens, the detection of mycoplasma-derived nucleases has potential clinical diagnostic applications. DNase activity has in fact been used to differentiate among various bacterial pathogens, including the identification of coagulase-positive staphylococci, such as Staphylococcus aureus, in bovine milk samples (Elston and ELSTON, 1968; Boerlin et al., 2003). Such assays depend on the isolation of the bacteria from biological fluids and tissues that also contain DNases, which would otherwise generate background; isolation and culture of the bacteria can consume valuable time. The use of chemically modified nucleic acids provides an alternative that could be used in more clinically relevant settings without generating background. Indeed, we developed a chemically modified RNase substrate that rapidly and robustly detected mycoplasma-derived ribonuclease activity in serum-containing conditioned media; digestion of this RNase substrate in the same media conditioned by an uncontaminated culture was minimal. Chemically modified nucleic acids can thus facilitate the rapid determination of the presence of bacterial contamination.
The particular nuclease detection assays we developed here may have limited specificity with respect to the type of bacterial contamination, because the chemically modified RNA oligos used here are likely to be susceptible to rapid degradation by nucleases produced by a variety of bacterial species (Moore et al., 2011). Interestingly, there are many nucleic acid modifications that have been developed that have unknown susceptibilities to bacterial nuclease degradation (Venkatesan et al., 2003). It is conceivable that future studies with additional chemically modified RNase substrates could reveal substrate-specific signatures of particular bacteria-derived nucleases that would facilitate the rapid identification of bacterial pathogens for clinical diagnostic purposes.
Footnotes
Acknowledgments
The authors would like to thank Dr. Paloma H. Giangrande and members of her lab for helpful discussions and Laynez W. Ackermann for technical help preparing M. fermentans cultures. This work was supported in part by the National Institute of Allergy and Infectious Diseases [AI083211 to ARH].
Author Disclosure Statement
No competing financial interests exist.