
Abstract
A manufacturing and purification process for duplex oligonucleotides was established, which shortens and simplifies currently used procedures, yielding a product of higher purity. The reported procedure is based on nondenaturing anion-exchange (AEX) chromatography, which is performed on the annealed duplex rather than the individual single strands. The duplex is formed early in the process by annealing of the crude single strands directly after solid-phase synthesis. Two 30 μmol manufacturing runs using duplex purification were performed on 2 different AEX resins and compared with a manufacturing run of the same scale using conventional single-strand chromatography. The same pooling strategy was employed for all purifications. Content of optimal duplex (duplex exclusively comprising full-length single strands) was 90.5% and 90.2% for the batches obtained by duplex purification and 86.1% for the batch obtained by single-strand purification. Maximum chromatographic recoveries were 67% for the duplex purification and 68% for the single-strand purification. Hence, the manufacture of small interfering RNA (siRNA) using duplex purification was simpler and faster than conventional single-strand purification and provided better purity and similar yield of final siRNA.
Introduction
Synthetic siRNA molecules are formed by 2 at least partially complementary RNA single strands, namely, the passenger strand and the guide strand. Typical strand lengths are 19–23 nucleotides. Strand association is mediated noncovalently via base-pairing and stacking interactions. Single strands are chemically synthesized stepwise from the 3′ to the 5′ terminus, employing conventional solid-phase phosphoramidite chemistry (Beaucage and Iyer, 1992; SPROAT, 2005). After chemical synthesis, the strands are cleaved from the support and deprotected. Subsequent chromatographic purification and desalting yield the purified single strands (Warren and Vella, 1995; Deshmukh et al., 2000; SHANAGAR, 2005). The final siRNA duplex is formed by mixing the 2 purified single strands in near equimolar ratio, typically followed by a heating and cooling phase (i.e., annealing) to promote the formation of the thermodynamically most stable duplexes. The resulting siRNA usually contains duplex molecules, including optimal duplex (2 full-length single strands) and nonoptimal duplex variants (containing at least 1 single strand impurity and/or mismatched sequences) as well as nonhybridized single strands.
Chromatographic purification of single strands before annealing to yield the final duplex is performed in the manufacture of virtually all therapeutic duplex oligonucleotides (Srivatsa, 2011) (see Fig. 1, left panel). The employed chromatography methods are well established, are scalable, and offer high resolution and many applications have been described using anion exchange (AEX) as well as reversed-phase techniques (Warren and Vella, 1995; Huber and OBERACHER, 2001; Cramer et al., 2011; Thayer et al., 2011). AEX in particular has been used in analytical and preparative applications for nucleic acids for many years [reviewed in ref. (Warren and Vella, 1995)]. To avoid the formation of stable secondary structures, denaturing conditions are typically used in chromatographic separations of oligonucleotides (Bourque and Cohen, 1994; Srivatsa et al., 1997; Deshmukh et al., 1998; SHANAGAR, 2005; Thayer et al., 2010). Nondenaturing applications are less common and are usually employed to determine the strand ratio of passenger and guide strand of siRNA after annealing (Thayer et al., 2011). Notwithstanding its success in the manufacture of duplex oligonucleotides, there are some inherent flaws in the conventional purification procedures. First, performing chromatography on both single strands, instead of purifying the duplex, doubles the requirements for reagents, buffer solutions, time, and workload. Second, to avoid a large excess of 1 strand over the other, an elaborate titration process of the 2 individual single strands is required before annealing, further increasing workload and process time. Finally, as the purification step is performed on the single-strand intermediates rather than the final duplex, the remaining single- and double-stranded impurities are not removed from the final duplex. This is particularly important, because the elevated temperatures employed during purification and annealing can lead to significant process-related strand degradation (Seiffert et al., 2011). Process-related impurities contaminating a drug substance are believed to convey risks and should always be reduced to the lowest levels that are reasonably practical (Jacobson-Kram and McGovern, 2007).

Flow schemes of small interfering RNA (siRNA) manufacturing. Left scheme: siRNA manufacturing using single-strand purification. Right scheme: siRNA manufacturing using duplex purification.
The main reason why purification of siRNA is performed on the single-strand intermediates, rather than the final duplex, is the fact that chromatographic separation of the hybridized duplex by nondenaturing methods is challenging. Current understanding purports that, because of almost identical physicochemical properties of optimal duplex and nonoptimal duplex variants, chromatographic separation between these species is difficult to achieve. However, several nondenaturing applications have been reported for AEX high-performance liquid chromatography (HPLC) (Wincott et al., 1995; Sproat et al., 1999; Thayer et al., 2007) and ion-pair reversed-phase (IP-RP) HPLC (Premstaller et al., 2000; Oberacher et al., 2001; Gelhaus et al., 2003; Beverly et al., 2005; Waghmare et al., 2009). High-resolution separations have been demonstrated for the separation of optimal duplex (full-length single strands only) from nonoptimal duplex variants (containing shortmers, longmers, and 2’,5’-isomers) using IP-RP HPLC under nondenaturing conditions (Beverly et al., 2005; Beverly et al., 2006; McCarthy et al., 2009; Noll et al., 2011; Seiffert et al., 2011). A very elegant method that solved the problem of duplex separation for siRNA has been reported by McCarthy et al. (2009), who combined duplex purification with on-column annealing using IP-RP HPLC. Notwithstanding its usefulness in analytical applications, large-scale purification by IP-RP HPLC has a number of drawbacks. Compared with AEX resins, reversed-phase resins have only moderate loading capacity. Cost of mobile-phase components and separation matrices are considerably higher. In addition, some organic solvents and ion-pairing reagents are difficult to remove from the purified duplex solution after IP-RP chromatography and may even require further chromatographic purification. Hence, oligonucleotide purification during mid- or large-scale manufacturing is typically performed using AEX rather than IP-RP chromatography.
Considering the good separations that have been achieved for oligonucleotide duplexes in analytical nondenaturing AEX, it is somewhat surprising that no preparative nondenaturing AEX chromatography method for oligonucleotide duplexes has been yet described. A manufacturing process for siRNA that involves purification of the final duplex using nondenaturing AEX chromatography is expected to offer many advantages over single-strand AEX purification and duplex IP-RP purification (see Fig. 1, right panel). Here, we investigated nondenaturing AEX chromatography as the main purification step of the hybridized duplex.
Materials and Methods
Chemicals
Acetonitrile (ACN; LC-MS Chromasolve grade), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; puriss. p.a.), hexylamine (puriss., GC grade), and triethylamine (puriss. p.a.) were purchased from Fluka (Sigma-Aldrich, St. Louis, MO). Phosphate-buffered saline (PBS; 10×, pH 7.4) was purchased from GIBCO (Life Technologies, Carlsbad, CA). A Milli-Q 185plus apparatus (Millipore, Bedford, MA) was used to prepare deionized water (>18 MΩ·cm) for all solutions and mobile phases.
RNA
All oligonucleotides were synthesized by conventional solid-phase synthesis. siRNA-Luc is a double-stranded RNA formed by annealing of full-length passenger strand (PS-Luc) and full-length guide strand (GS-Luc). The guide strand sequence is complementary to a region in the mRNA of firefly luciferase. Sequences of PS-Luc and GS-Luc are listed in Table 1; the duplex is depicted in Supplementary Fig. S1 (Supplementary Data are available online at www.liebertpub.com/nat). Base composition of full-length passenger strands and full-length guide strands of siRNA-Luc and siRNA-1 to siRNA-4 are listed in Table 2. All oligonucleotides were manufactured at Roche Kulmbach GmbH.
Mr indicates molecular weight. Uppercase letters indicate 2′-OH RNA nucleotides; lower case letters indicate 2′-O-methyl nucleotides; TT indicates two 2′-deoxy-thymidine nucleotides connected via a phosphorothioate linkage.
A cyclic phosphate at the 3′ end.
PS-Luc, full-length passenger strand of siRNA-Luc; GS-Luc, full-length guide strand of siRNA-Luc; siRNA, small interfering RNA.
Uppercase letters indicate 2′-OH RNA nucleotides; lowercase letters indicate 2′-O-methyl nucleotides; TT indicates two 2′-deoxy-thymidine nucleotides connected via a phosphorothioate linkage.
Annealing conditions
If not stated otherwise, equimolar amounts of passenger and guide strands were annealed by mixing, heating to 85°C for 10 minutes followed by cooling to room temperature over a period of 3 hours. The employed extinction coefficients were 211.9 OD (OD is optical density at 260 nm, 1 OD equals approximately 40 μg of RNA)/μmol and 200.9 OD/μmol for GS-Luc and PS-Luc, respectively. Extinction coefficients were calculated using the nearest neighbor model as described by Cavaluzzi and Borer (2004). Analytical samples were 50 μM siRNA in 1× PBS. About 30 μmol samples were 493 μM siRNA-Luc in water. Until analysis, all solutions were stored at +2°C to +8°C.
AEX method development
AEX purifications during method development were carried out on a Dionex Ultimate 3000 series HPLC system (Dionex, Sunnyvale, CA) using UV detection at 260 nm. Oligonucleotides were separated by salt-gradient elution on a Mini Q 4.6×50 mm column with a 5 μm bead size (GE Healthcare, Pollars Wood, United Kingdom) or a TSKgel SuperQ-5PW 7.5×75 mm column with a 10 μm bead size (Tosoh Biosciences, King of Prussia, PA). Mobile phases contained 20 mM sodium phosphate at pH 6.5, 7, or 8. Mobile phase B contained 1 M sodium chloride or sodium bromide. The gradient was run from 20% to 60% of mobile phase B in 25 minutes at a flow rate of 1 mL/minute for the Mini Q column and 2 mL/minute for the TSKgel SuperQ-5PW column. Column temperature was 40°C, 50°C, or 60°C. Injection volume was 2 μL of a 50 μM RNA solution in 1× PBS.
Analytical AEX
Fraction analysis was carried out on a Dionex Ultimate 3000 series HPLC system (Dionex) using UV detection at 260 nm. Fractions were separated by salt-gradient elution on a DNAPac-200 4×250 mm column (Dionex). Mobile phases A and B contained 25 mM Tris (pH 8) and 10% ACN. Mobile phase B contained 800 mM NaClO4. The gradient was run from 10% to 28% of mobile phase B in 12 minutes at a flow rate of 1 mL/minute. Column temperature was 75°C. All fractions were adjusted to 1 OD/mL (5 ng/mL) with water; injection volume was 20 μL.
Preparative AEX (30 μmol)
Duplex AEX purification was carried out on an ÄKTA Purifier HPLC system (GE Healthcare) using UV detection at 260 nm. siRNA-Luc duplexes were separated on a 5×100 mm column containing 2 mL Source 15Q resin (GE Healthcare) or a 5×100 mm column containing 2 mL TSKgel SuperQ-5PW (Biosciences, King of Prussia, PA). Mobile phases A and B contained 20 mM sodium phosphate (pH 6.5) and 10% ACN. Mobile phase B contained 1 M sodium bromide. The gradient was run from 10% to 50% of mobile phase B in 64 minutes at a flow rate of 0.9 column volume (CV)/minute; column temperature was 60°C.
Single-strand AEX purification was carried out on an ÄKTA Purifier HPLC system (GE Healthcare) using UV detection at 260 nm. GS-Luc or PS-Luc was separated on a 5×100 mm column containing 2 mL Source 15Q resin (GE Healthcare). Mobile phases A and B contained 20 mM sodium phosphate (pH 7.8). Mobile phase B contained 1 M sodium bromide. The gradient was run from 10% to 50% of mobile phase B in 109 minutes at a flow rate of 0.2 CV/minute. Column temperature was 45°C. Chromatographic yield was determined by comparing total loaded RNA to total RNA in the pooled fractions.
Desalting
Crude single strands as well as pool fractions of the AEX purifications were desalted using a Sephadex HiPrep 26/10 desalting column (GE Healthcare). Desalting was performed on an ÄKTA Purifier HPLC system (GE Healthcare) using UV detection at 260 nm. Mobile phase was water; flow rate was 10 mL/minute.
Size-exclusion chromatography
Size-exclusion chromatography (SEC) was carried out on a Dionex Ultimate 3000 series HPLC system (Dionex) using UV detection at 260 nm. Oligonucleotides were separated on a Superdex 75 column, 10/300 GL (GE Healthcare) at room temperature. Mobile phase consisted of 1×PBS containing 10% ACN. The flow rate was 0.014 CV/minute. Injection volume was 5 μL of a 50 μM RNA solution in 1× PBS.
Nondenaturing IP-RP HPLC
For nondenaturing IP-RP HPLC separations, samples were applied to an Ultimate 3000 RS series HPLC system (Dionex) using UV detection at 260 nm. Oligonucleotides were separated by ACN-gradient elution on an Acquity UPLC OST C18 2.1×100 mm column with a 1.7 μm bead size (Waters, Milford, MA). Mobile phase A consisted of 20 mM HA, 100 mM HFIP, and 5% ACN. Mobile phase B consisted of 80% ACN. The gradient was run from 15% to 35% of mobile phase B in 25 minutes at a flow rate of 250 μL/minute. Injection volume was 2 μL of a 50 μM RNA solution in 1× PBS. Column temperature was 20°C.
Denaturing IP-RP HPLC
For denaturing IP-RP HPLC separations, samples were applied to an Ultimate 3000 RS series HPLC system (Dionex) using UV detection at 260 nm wavelength. Oligoribonucleotides were separated by methanol-gradient elution on an XBridge C18 OST 2.1×50 mm column with a 2.5 μm particle size (Waters). Mobile phase A consisted of 100 mM HFIP, 16.3 mM TEA, and 1% methanol. Mobile phase B consisted of 100 mM HFIP, 16.3 mM TEA, and 95% methanol. Column temperature was 65°C. A gradient was run from 1% to 18% of mobile phase B in 30 minutes at a flow rate of 250 μL/minute. Injection volume was 2 μL of a 50 μM RNA solution in water.
Mass spectrometry
For mass spectrometric analysis, an LCQ Deca XP+Ion-Trap mass spectrometer equipped with an ESI interface (ThermoFisher Scientific, Waltham, MA) was used in line with the IP-RP HPLC system. The mass spectrometer was run in negative ion mode, spray voltage was 4.5 V, capillary temperature was 315°C, capillary voltage was −100 V, and scanning mass ranged from 600 to 2000 m/z. Data acquisition and analysis was performed using the Xcalibur software as well as the ProMass deconvolution software (ThermoFisher Scientific).
Results and Discussion
AEX method development
Chromatography conditions and resins for nondenaturing AEX duplex purification were established by performing a series of chromatographic runs at analytical scale. To allow for later process scale-up, buffer components and resins compatible with mid- and large-scale manufacturing were employed. Both the investigated AEX resins, Source 15Q and TSKgel SuperQ-5PW, offer high loading capacity and are available in bulk quantities. They utilize the same functional group, a quaternary amine, but differ in matrix chemistry. Source Q material consists of polystyrene/divinyl benzene beads. The TSKgel SuperQ-5PW beads are made from poly(methyl-methacrylate). Development runs were performed at analytical scale using MiniQ columns with a bead size of 5 μm and TSKgel SuperQ-5PW columns with bead sizes of 10 μm. The MiniQ column was employed, because the 15 μm beads of Source 15Q resin was not designed to operate optimally at analytical flow rates. Initial scouting runs were performed using phosphate buffer systems at neutral, slightly acidic, or basic pH (pH 7, 6.5, or 8), using sodium bromide or sodium chloride as eluting agents. To maintain nondenaturing properties, column temperature did not exceed 60°C, which is >20°C below the melting temperature of all tested siRNAs. For separations of siRNA-Luc, sodium bromide provided better results than sodium chloride on both columns. On the MiniQ column, separations were best at the lower pH (pH 6.5), whereas performance of the TSKgel SuperQ-5PW column was best at pH 8 (data not shown). Separation between single and double strands of siRNA-Luc were slightly better using the TSKgel SuperQ-5PW column compared with the Source 15Q column (Table 3).
Mobile phases contained 20 mM sodium phosphate at pH 6.5. Mobile phase B contained 1 M sodium bromide. The gradient was run from 20% to 60% of mobile phase B in 25 minutes at a flow rate of 1 or 2 mL/minute, respectively. Column temperature was 40°C, 50°C, or 60°C. RRTs of the excess single strands are indicated relative to the main duplex peak.
No=no separation between peaks was observed.
RRT, relative retention time; ACN, acetonitrile.
In all following experiments, mobile phases containing sodium bromide were used at pH 6.5, either with or without ACN as organic modifier. Five different siRNAs (siRNA-Luc and siRNA-1 to siRNA-4) containing excess of either passenger or guide strand were analyzed. This set of siRNAs encompassed 5 passenger strands and 4 guide strands of different base composition and modification patterns (including 2’-deoxy and 2’-OMe sugar modifications, and phosphorothioate backbone modifications) and were intended to represent a diverse array of siRNA molecules (Table 2). Despite some differences between the retention behavior of individual siRNAs, some general conclusions could be drawn. Of the 3 column temperatures evaluated (40°C, 50°C, and 60°C), the highest temperature gave the best separations on both columns. This is in accordance with the reported performance of AEX columns for polyanions (Thayer et al., 2010, 2011). Baseline separation between double and both single strands was achieved for all 5 siRNAs on the MiniQ column and for 3 of 5 siRNAs on the TSKgel SuperQ-5PW column (Table 3).
In the analyses of all siRNA, a remarkable difference in the elution profiles of the 2 columns was observed. On the MiniQ column, single strands eluted earlier than the duplex, whereas on the TSKgel SuperQ-5PW column this elution order was reversed (Fig. 2). We attributed this difference primarily to nonelectrostatic interactions of duplex and single strands with the resin matrix. Although AEX separation relies primarily on electrostatic interactions between the charged phosphates of the oligonucleotide backbones and the functional groups of the stationary phase, secondary mode interactions (such as hydrophobic Van der Waals interactions) between the matrix surface of the column and the hydrophobic bases of the nucleotides are possible (Thayer et al., 2011). Single-stranded and duplex oligonucleotides differ significantly in their ability to interact with the surfaces of the column resins. In general terms, a single strand may be considered a more flexible structure, where the π-electron systems of the bases have a certain ability of participating in hydrophobic interactions with surfaces. In contrast, the helical double strand is considered more rigid and has rod-like shape. The charged phosphates are positioned at the outside of the Watson–Crick helix, and the heterocyclic bases, connected via hydrogen bonds, form the hydrophobic core and may not be readily accessible for interactions with external surfaces. In separations employing the Source Q resin, single strands bound less tightly to the stationary phase, eluting earlier than the double strands. On the TSKgel SuperQ-5PW resin, single strands eluted later than the double strands, suggesting that secondary (hydrophobic) interactions with the column material contributed more strongly to the retention of the single strands than on the Source Q resin.
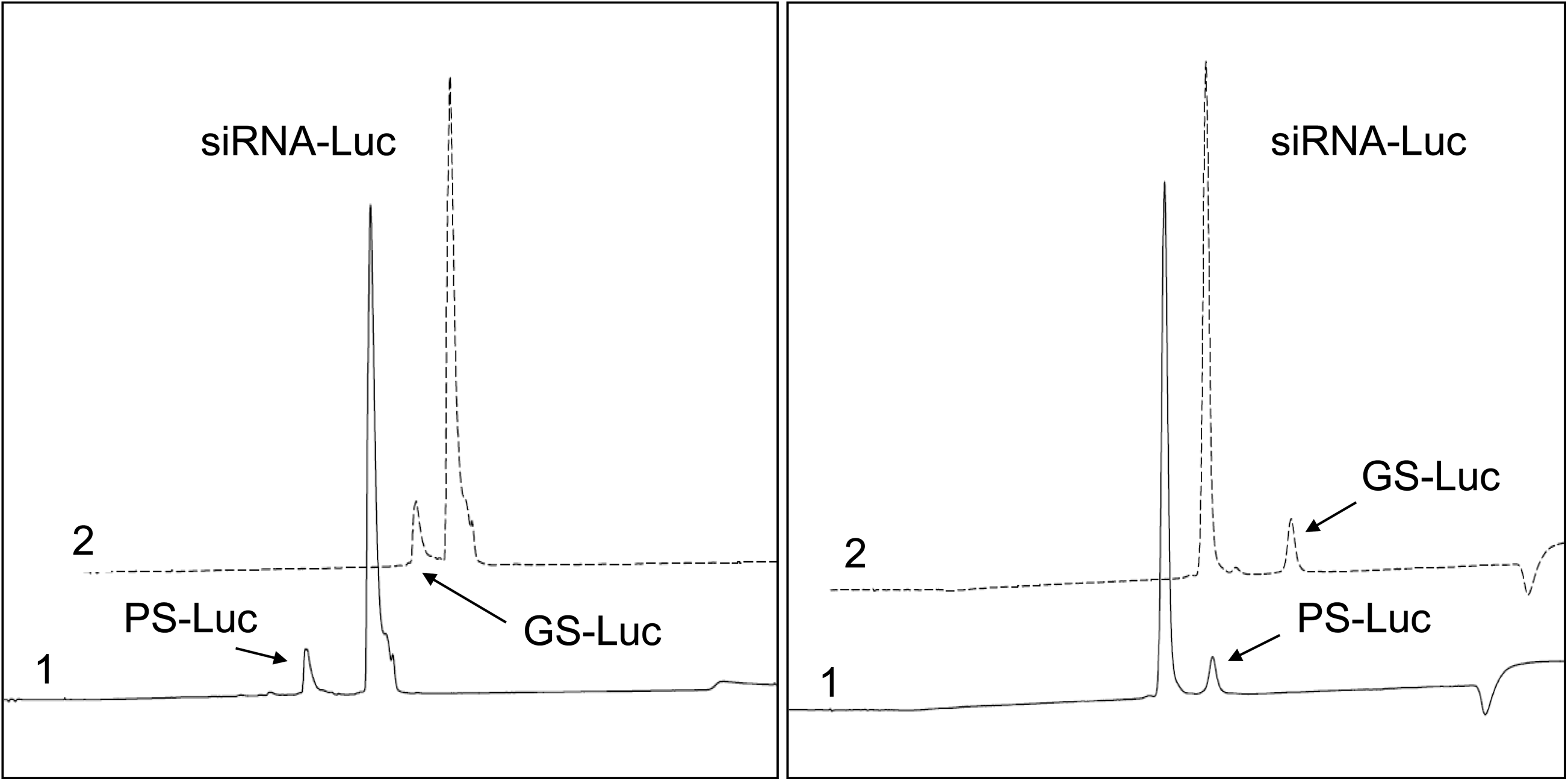
AEX scouting purifications. Chromatograms of siRNA-Luc spiked with PS-Luc (trace 1) and GS-Luc (trace 2). Separations were performed using MiniQ columns (left panel) and TSKgel SuperQ-5PW columns (right panel). Mobile phases A and B contained 20 mM sodium phosphate at pH 6.5. Mobile phase B contained 1 M sodium bromide. The gradient was run from 20% to 60% of mobile phase B in 25 minutes. Flow rate was 1 mL/minute for the MiniQ column and 2 mL/minute for the SuperQ-5PW column. Column temperature was 60°C. AEX, anion exchange; PS-Luc, full-length passenger strand of siRNA-Luc; GS-Luc, full-length guide strand of siRNA-Luc; siRNA, small interfering RNA.
Preparation of the duplex batches
Desalted solutions of crude GS-Luc and crude PS-Luc were adjusted to 1.5 μmol/L in water. Concentration measurements based on the theoretical extinction coefficients of the strands were used (see Materials and Methods). Purity of the crude strands was 75.5% for GS-Luc (Supplementary Fig. S2, upper trace) and 75.8% for PS-Luc (Supplementary Fig. S3, upper trace) as measured using denaturing IP-RP. Equimolar amounts of the solutions were mixed [1.563 mL of GS-Luc (316.4 OD) and 1.354 mL of PS-Luc (300 OD)]; the resulting solution was heated to 85°C for 10 minutes and allowed to cool to room temperature over a period of 3 hours. Total duplex content by SEC was almost identical before and after annealing (Table 4, Fig. 3, upper panels). However, in nondenaturing IP-RP measurements, optimal duplex increased from 70.1% to 73.5%, whereas late-eluting duplexes decreased from 7.5% to 5.7%. Only small changes were recorded in the peak areas of the shortmer duplexes and the nonhybridized fraction (Fig. 3, lower panels). The recorded changes in duplex composition support the beneficial effect of thermodynamically controlled annealing. This confirms the initial assumption that in a mixture that contains single-strand impurities, an annealing process performed under thermodynamic control promotes redistribution of shortmers between duplexes and is required to obtain a maximal amount of optimal duplex.

Nondenaturing analysis of the crude single-strand mixture before and after annealing. Equimolar amounts of desalted crude GS-Luc (316.4 OD) and crude PS-Luc (300.0 OD) were mixed and analyzed using SEC (upper panels) and nondenaturing IP-RP (lower panels). Chromatography was performed before (straight line) and after (dashed line) annealing (85°C, 10 minutes). Full chromatograms are depicted in the left panels; enlarged chromatograms are depicted in the right panels. SEC, size-exclusion chromatography; IP-RP, ion-pair reversed phase. 1 OD equals approximately 40 μg RNA.
Total duplex content was determined by SEC; optimal duplex content was determined by nondenaturing IP-RP.
SEC, size-exclusion chromatography; IP-RP, ion-pair reversed phase.
For preparative duplex purifications, chromatographic conditions that allow for earlier elution of nonhybridized single strands, compared with the duplex, are preferred. In this case, nonhybridized shortmers are less likely to co-elute with the intact duplex. However, as very good separation between single and double strands was achieved for both columns, Source 15Q and TSKgel SuperQ-5PW, both matrix materials, were evaluated for the preparative purification of siRNA-Luc. Chromatographic conditions of the preparative runs were based on the results of the AEX development runs. Mobile phase contained phosphate buffer (pH 6.5), 10% ACN, and sodium bromide as eluting agent. Flow and gradient conditions were scaled from the AEX development runs. Source 15Q material with a bead size of 15 μm and TSKgel SuperQ-5PW resin with a bead size of 20 μm were employed. About 2 mL of resin was used in columns of 100 mm length. Flow rates were 0.9 CV/minute for both columns. Column temperature was 60°C. Annealed crude strands with 635 OD (approximately 12.7 mg RNA per gram resin, Batch A) and 616 OD (approximately 12.3 mg RNA per gram resin, Batch B) were applied on the Source 15Q and TSKgel SuperQ-5PW columns, respectively. Fractions were collected and purity of the fractions was determined by nondenaturing IP-RP (Source 15Q: Figs. 4 and 5; TSKgel SuperQ-5PW: Figs. 6 and 7). Fractions with a content of optimal duplex of 85% or higher were combined. The combined fractions constituted the final duplex batches for Source 15Q (Batch A) and TSKgel SuperQ-5PW (Batch B) purifications (Tables 5 and 6). Batches were analyzed using SEC (Supplementary Fig. S4) and denaturing and nondenaturing IP-RP (Figs. 8 and 9). Total duplex content determined by SEC was 99.2% for Batch A and 99.3% for Batch B. Optimal duplex content as determined by nondenaturing IP-RP was 90.5% for Batch A and 90.2% for Batch B (Table 4). Purification of 635 OD crude siRNA-Luc using the Source 15Q resin yielded 318 OD purified duplex, which corresponds to a total recovery from chromatography of 66.7%, calculated from the amount of the applied crude strands. Purification of 616 OD crude siRNA-Luc using TSKgel SuperQ-5PW resin yielded 275 OD purified duplex, which corresponds to a total recovery from chromatography of 59.2%.

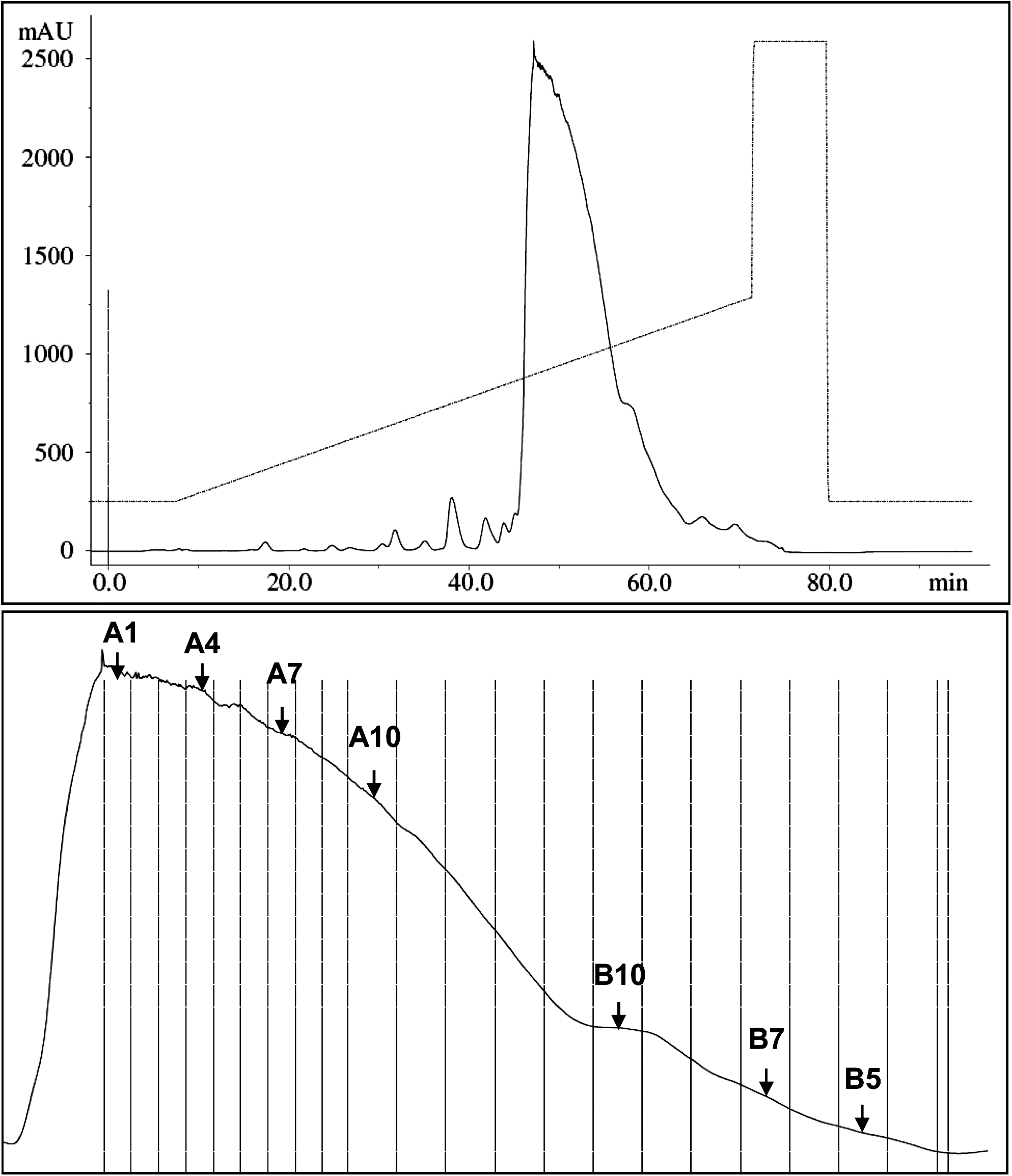
Preparative duplex chromatography using Source 15Q. The 635 OD siRNA-Luc was separated using 2 mL of Source 15Q resin. Upper panel depicts full-sized curve with salt gradient. Lower panel depicts the enlarged duplex peak section with fraction markers. Selected fractions are indicated by arrows. Fractions A7 to A12 and B5 to B12 were combined to yield the final Batch A.
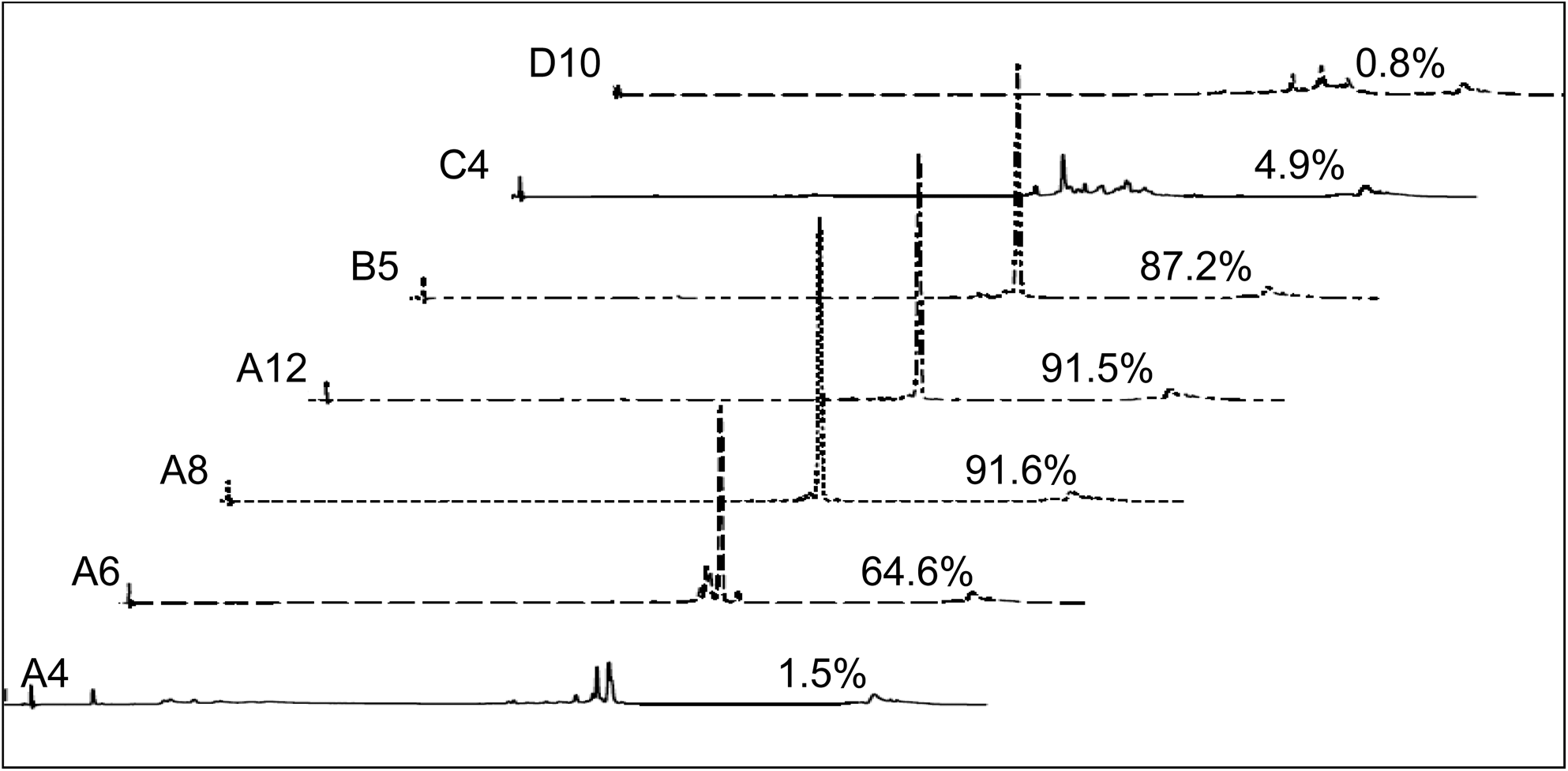
Nondenaturing IP-RP analysis of selected fractions of the Source 15Q duplex purification (Batch A). Optimal duplex contents of the fractions are indicated above the respective chromatography trace. Fractions A7 to A12 and B5 to B12 were combined to yield the final Batch A.
Preparative AEX duplex chromatography using a TSKgel SuperQ-5PW column. The 616.4 OD siRNA-Luc was separated using 2 mL of TSKgel SuperQ-5PW resin. Upper panel depicts full-sized curve with salt gradient. Lower panel depicts the enlarged duplex peak section with fraction markers. Selected fractions are indicated by arrows. Fractions A2 to A11 were combined to yield the final Batch B.
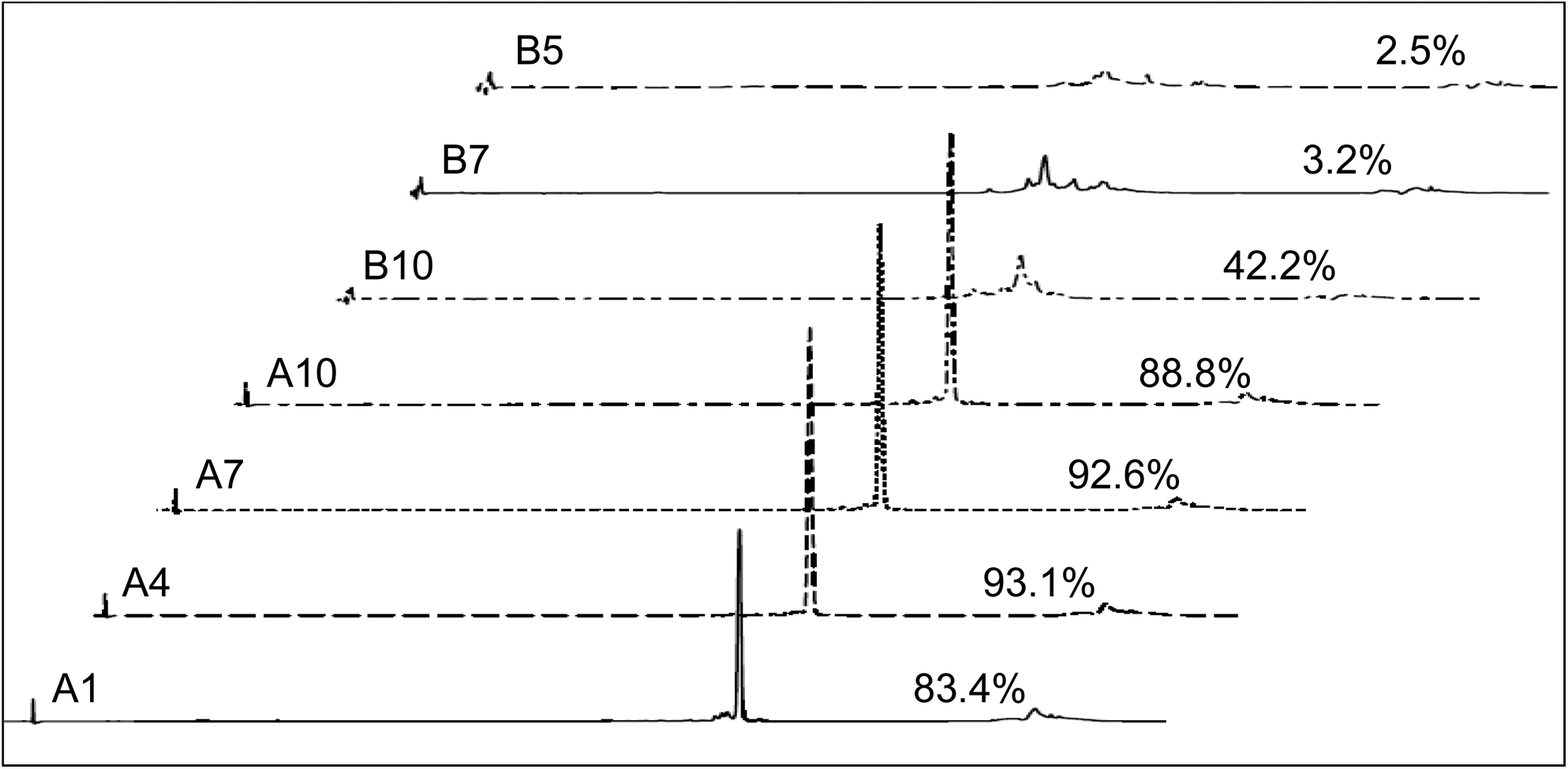
Nondenaturing IP-RP analysis of selected fractions of the TSKgel SuperQ-5PW duplex purification (Batch B). Optimal duplex contents of the fractions are indicated above the respective chromatography trace. Fractions A2 to A11 were combined to yield the final Batch B.

Denaturing IP-RP analysis of final single-strand and duplex purification batches. Overlay of the full chromatograms of Batch A (straight line), Batch B (dashed line), and Batch C (dotted line) is depicted in the upper panel. Peak areas of GS-Luc and PS-Luc are indicated. The lower panel depicts an overlay of the enlarged peak areas. Peak i=5’(N-5)GS; peak ii=5’(N-4)PS; peak iii=5’(N-8)PS.
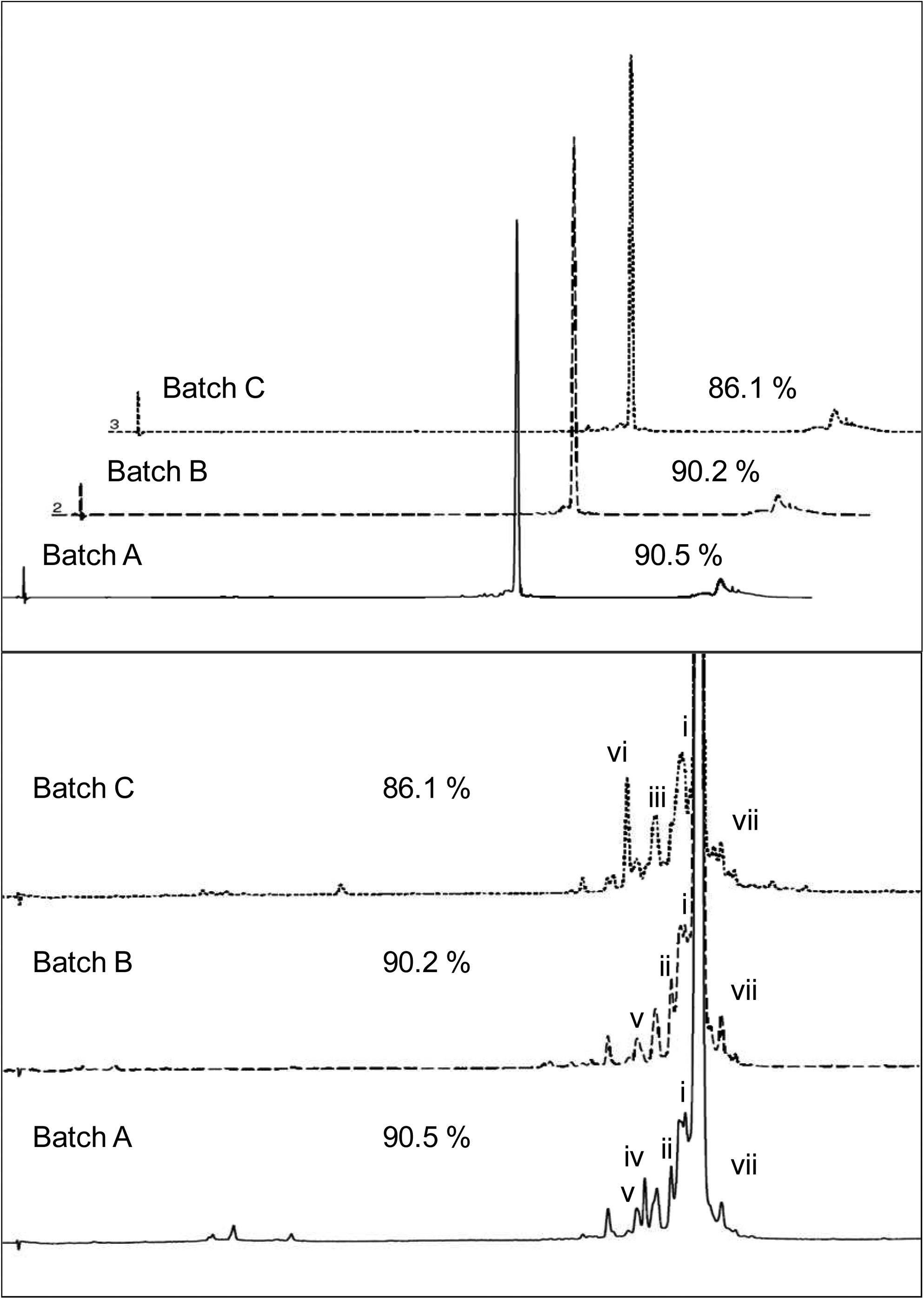
Nondenaturing IP-RP analysis of final single-strand and duplex purification batches. Overlay of the full chromatograms of Batch A (straight line), Batch B (dashed line), and Batch C (dotted line) is depicted in the upper panel. Peak areas of siRNA-Luc optimal duplex are indicated. The lower panel depicts an overlay of the enlarged peak areas. Peak i=duplexes containing shortmers PO-GS, PO-PS, and 5’(N-1)GS; peak ii=duplex 5’(N-2)GS/PS-Luc; peak iii=GS-Luc/5’(N-1)PS and GS-Luc/5’(N-2)PS; peak iv=5’(N-4)GS/PS-Luc and 5’(N-5)GS/PS-Luc; peak v=3’(N-3)GS*/PS-Luc and GS-Luc/5’(N-8)PS; peak vi=GS-Luc/5’(N-4)PS; peak vii=duplexes containing longmers.
Peak areas of the optimal duplex are expressed in percentage as determined by nondenaturing IP-RP analysis. Fractions A7 to A12 and B4 to B12 were combined to yield the final Batch A. Pooled fractions are indicated by bold letters.
Peak areas of the optimal duplex are expressed in percentage as determined by nondenaturing IP-RP analysis. Fractions A2 to A11 were combined to yield the final Batch B. Pooled fractions are indicated by bold letters.
Preparation of the single-strand batch
Chromatographic purification of the crude GS-Luc and PS-Luc single strands was performed using the Source 15Q column. To resemble typical purification conditions, chromatographic parameters were selected analogous to a regularly used in-house purification process. Mobile phase contained phosphate buffer (pH 6.5) and sodium bromide as eluting agent. Fractions of the single-strand chromatography were collected and purity of the fractions was determined by AEX (data not shown). Fractions with purities of 85% or higher were included in the respective single-strand pools. Purification of 635 OD crude GS-Luc (purity=75.5) yielded 416 OD of purified strand (purity=93.7%), which corresponds to a chromatographic recovery of 65.5%, calculated from the amount of the applied crude strand (Supplementary Fig. S2). Purification of 635 OD crude PS-Luc (Purity=75.8%) yielded 432 OD of purified strand (Purity=91.9%), which corresponds to a chromatographic recovery of 68% (Supplementary Fig. S3). Equimolar amounts of the purified single strands were annealed (300 OD PS-Luc and 316 OD GS-Luc) by heating to 85°C for 10 minutes and cooling to RT over a period of 3 hours. Optimal duplex content of the resulting siRNA-Luc batch (Batch C) was 86.1%, as determined by nondenaturing IP-RP (Table 4).
Batch comparison
The final siRNA-Luc batches were compared using the results from SEC, denaturing IP-RP, and a previously reported, high-resolution nondenaturing IP-RP method (Noll et al., 2011). Both IP-RP methods were used in combination with ESI-MS detection. In the analyses, Batches A and B (prepared by duplex chromatography) showed superior purity than Batch C (prepared by single-strand chromatography). The difference is particularly obvious for optimal duplex content, which was >4% higher in the duplex purified batches compared with the single-strand purified batch (Table 4). Duplex impurities containing shortmers (PO-GS, PO-PS, 5’(N-1)GS; peak i, Fig. 9) were higher in Batch C (5.8%) compared with Batch A (3.6%) and Batch B (4.7%). The major nonoptimal duplex detected in Batch A was a peak of 0.6% total peak area (peak iv, Fig. 9, lower trace), containing 5’(N-5)GS/PS-Luc and 5’(N-4)GS/PS-Luc. These 2 nonoptimal duplexes were not detected in Batch B and Batch C (Fig. 9, middle and upper trace). The major nonoptimal duplex detected in Batch C was 5’(N-4)PS/GS-Luc (peak vi, Fig. 9, upper trace). The amount was 1.3% of total peak area. In Batch A and Batch B, this nonoptimal duplex was detected as a minor component of peak v (<0.3% in both batches; Fig. 9). The difference in the amount of 5’(N-4)PS in all 3 batches is visible in the denaturing chromatograms (peak ii, Fig. 8).
Twice the amount of duplex impurities containing longmers were detected in Batch C (2.8%; peaks vii, Fig. 9, upper trace) compared with Batch A (1.3%) and Batch B (1.4%). Longmers were guide strand with an additional guanine (peak vii, Fig. 9, lower and middle trace) in all 3 batches. Additional longmers containing additional U or C bases as well as CNET longmers (peaks vii, Fig. 9, upper trace) were detected in Batch C. These results were supported by denaturing IP-RP (Fig. 8). The major impurities detected in the 3 batches are listed in Table 1.
Only small amounts of nonhybridized single strands were detected in all 3 batches. In Batch A and Batch C, nonhybridized strands contained PS-Luc and shortmers as small as N-6 of both GS-Luc and PS-Luc. Batch A predominantly contained nonhybridized 5’(N-5)GS and 5’(N-4)GS. The 5’(N-5)GS impurity was quantified to 0.4% total peak area by denaturing IP-RP (peak i, Fig. 8, lower trace). Much lower amounts of this impurity were detected in Batch B and Batch C, which corresponds well to the results from nondenaturing analysis (peak iv, Fig. 9, upper and middle trace). Batch B contained 0.2% of nonhybridized 5’(N-8)PS shortmer, which was also detected in the denaturing IP-RP (peak iii, Fig. 8, middle trace, 0.3%). The presence of this shortmer was attributed to co-elution of the shortmer with the siRNA-Luc duplex during duplex purification, because on the TSKgel SuperQ-5PW resin full-length single strands eluted after the duplex peak.
Taken together, higher content of optimal duplex, less longmer impurities and lower amounts of shortmer impurities were detected in Batches A and B compared with Batch C. Hence, using the same pooling strategy for all 3 batches, longmer as well as shortmer impurities were removed more efficiently by duplex chromatography compared with single-strand chromatography. These results confirm the original assumption that purification of the final duplex should allow for higher purity compared with a process wherein purification is performed on the single-strand intermediates.
Conclusion
In the work reported here, we have established a manufacturing process for duplex oligonucleotides that shortens and simplifies the currently used procedures, yielding a product of higher purity. The reported procedure is based on nondenaturing AEX purification, performed on the duplex rather than on both single strands individually. The same resins and mobile phase components were used as in conventional single-strand chromatography. The advantages of performing chromatographic purification on the siRNA duplex rather than on the single-strand intermediates are clearly at hand: Chromatographic steps during manufacture of siRNA are reduced from 2 to 1 and annealing is significantly simplified. Annealing is performed upstream of duplex purification and labor-intensive titration of the 2 strands is no longer required. Subsequent duplex purification allows for removal of most nonhybridized strands as well as nonoptimal duplexes and enables optimization of the chromatographic conditions on the siRNA drug substance.
Although the AEX duplex purifications (Batches A and B) represent our first experiments at the reported scale and may not have been completely optimized, optimal duplex contents of both batches prepared by duplex chromatography were significantly better than those of the batch prepared by single-strand chromatography (Batch C). At the same time, maximal chromatographic recovery from duplex chromatography was the same as for single-strand chromatography. Drawing on our extensive experience with manufacture of siRNA at 300–1200 μmol scale, the results of the 30 μmol single-strand purification (Batch C) were considered representative of a typical, high-quality preparation of siRNA using conventional manufacturing conditions. A general applicability of the duplex purification results (Batches A and B) is considered likely, because separation properties, as determined in the scouting experiments, were similar for 5 siRNAs of different sequences and modification patterns. It is reasonable to speculate that with increasing experience the reported duplex purification methods can be improved further.
Taken together, siRNA manufacture using nondenaturing AEX duplex purification was simpler and faster than the conventional single-strand purification and, using the same pooling strategy, provided similar yield and better purity of the final compounds. The reported method is useful for manufacturing of therapeutic double-stranded nucleic acids such as siRNA, miRNA, and decoy duplex DNA.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.