
Abstract
The toll-like receptors (TLRs) 7, 8, and 9 stimulate innate immune responses upon recognizing pathogen nucleic acids. Certain GU- or AU-rich RNA sequences were described to differentiate between human TLR7- and TLR8-mediated immune effects. Those single-stranded RNA molecules require endosomal delivery for stabilization against ribonucleases. We have discovered RNA sequences that preferentially activate TLR7, form higher ordered structures, and do not require specific cellular delivery. In addition, a dual activation of TLR8 and TLR9 without affecting TLR7 can be achieved by chimeric molecules consisting of GU-rich RNA and Cytosin (C) phosphordiester or phosphorthioat (p) guanine (CpG) motif DNA sequences. Such chimeras stimulate TLR9-mediated type I interferon (IFN) and TLR8-depending proinflammatory cytokine and chemokine production upon primary human cell activation. However, an RNA-dependent TLR7 IFN-α cytokine release is suppressed by the phosphorothioate DNA sequence contained in the chimeric molecule. To convert the immune response of a single-stranded RNA from TLR7/8 to TLR9, a simple chemical modification at the 5′ end proves to be sufficient. Such 8-oxo-2′-deoxy-guanosine or 8-bromo-2′-deoxy-guanosine modifications of the first guanosine in GU-rich single-stranded RNAs convert the immune response to include TLR9 activation and demonstrate strong additive effects for type I IFN immune responses in human primary cells.
Introduction
TLR7, TLR8, and TLR9 belong to the same subfamily of TLRs based on the genomic structure, sequence similarities, and endosomal localization (Chuang and Ulevitch, 2000). TLR9, TLR8, and TLR7 respond to specific nucleic acids or derivatives, and TLR7 and TLR8 are also stimulated by small molecules such as Resiquimod (Jurk et al., 2002). GU-rich single-stranded RNA from viruses or synthetic single-stranded oligoribonucleotides (ORNs) have been described as TLR7 and TLR8 ligands (Diebold et al., 2004; Heil et al., 2004; Hornung et al., 2004; Forsbach et al., 2007, 2008), whereas TLR9 recognizes nonmethylated, Cytosin (C) phosphordiester or phosphorthioat (p) guanine (CpG)-containing DNA of bacterial or viral origin, and synthetic CpG oligodesoxynucleotides (ODN) (Krieg et al., 1995; Heeg et al., 1998; Bauer et al., 2001). This subfamily has a unique pattern of cell type–specific expression that is thought to be responsible for different pathogen response profiles (Iwasaki and Medzhitov, 2004). TLR7 and TLR9 mRNAs are mainly detected in plasmacytoid dendritic cells (pDCs) and B cells, and TLR8 mRNA is observed in myeloid dendritic cells (mDCs), monocyte-derived dendritic cells (DCs), monocytes, macrophages, natural killer (NK) cells, and regulatory T cells (Krug et al., 2001a; Hornung et al., 2002; Ito et al., 2002; Zarember and Godowski, 2002; Kokkinopoulos et al., 2005; Peng et al., 2005). The target cell selectivity and diversity appears a useful tool, in addition with recombinant cells expressing these receptors, to differentiate in vitro between potential ligands for TLR7, TLR8, and TLR9.
ORNs activating TLR7/8 or only TLR7 or TLR8 have been already identified and contain specific RNA motifs (Heil et al., 2004; Hornung et al., 2005; Forsbach et al., 2007, 2008). For example, GU-rich ORN induce TLR7- and TLR8-mediated cytokine and chemokine release upon human immune cell activation, and AU-rich ORNs stimulate TLR8-dependent immune responses. However, all ORNs published so far require endosomal delivery to protect the RNA from endo- or exoribonuclease digestion. The aim of this study was first to discover ORNs activating the immune system that does not require endosomal delivery systems and second to generate new nucleic acid TLR ligands that combine TLR7, TLR8, and/or TLR9 immune responses. All studies were performed in human peripheral blood mononuclear cell (PBMC) and human stable transfected TLR7, TLR8, or TLR9 human embryonic kidney (HEK)-293 cells. Avoiding endosomal delivery systems do have the advantage of less toxicity and potential side effects. Our study design used different 3′ ORN modifications as well as nucleotide modifications to improve ORN stability and their protection against ribonucleases in the absence of a delivery system. Combined TLR7, TLR8, and/or immune response will have important impact in clinical development of several diseases like cancer or allergy. To explore this issue, we designed RNA-DNA chimeric oligonucleotides as well as 5′ chemically modified ORNs.
Materials and Methods
Reagents
ORNs and CpG ODN were provided by Coley Pharmaceutical GmbH. All substances were controlled for identity and purity by Coley Pharmaceutical GmbH and had undetectable endotoxin levels (<0.1 EU/mL) measured by the Limulus assay (BioWhittaker). Oligonucleotides were suspended in sterile endotoxin-free Tris-ethylenediaminetetraacetic acid (EDTA; CpG ODN; Sigma) or in DNAse- and RNAse-free water (ORN; Life Technologies), and were stored and handled under aseptic conditions to prevent contamination. 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) was obtained from Roche.
Peripheral blood mononuclear cells
PBMC preparations from healthy male and female human donors were obtained from the Institute for Hemostaseology and Transfusion Medicine of the University of Düsseldorf (Germany). All blood samples were given by voluntary donors for research purpose in a contract with the University of Düsseldorf. Buffy coats of those samples were bought via research contract from the University of Düsseldorf. PBMCs were purified by centrifugation over Ficoll-Hypaque (Sigma-Aldrich). Purified PBMCs were washed twice with 1×phosphate-buffered saline and resuspended in RPMI 1640 culture medium RPMI-1640 was developed by Moore et. al. at Roswell Park Memorial Institute, hence the acronym RPMI. The formulation is based on the RPMI-1630 series of media utilizing a bicarbonate buffering system and alterations in the amounts of amino acids and vitamins supplemented with 5% (v/v) heat inactivated human AB serum (BioWhittaker) or 10% (v/v) heat inactivated fetal calf serum, 1.5 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin (all from Sigma-Aldrich).
Cytokine and chemokine production
Freshly isolated PBMCs were resuspended at a concentration of 3×106/mL to 5×106/mL and added to 96-well round-bottomed plates (200 μL/well), which had previously received nothing or CpG ODN, ORN/CpG ODN chimera or ORN complexed to the indicated formulation or the formulation alone. Cells were cultured in a humidified incubator at 37°C for the indicated time points. Culture supernatants (SNs) were collected and, if not used immediately, were frozen at −20°C until required. Amounts of cytokines in the SN were assessed using commercially available enzyme-linked immunosorbent assay (ELISA) Kits (interferon γ [IFN-γ], interleukin 12p40 [IL-12p40], tumor necrosis factor α [TNF-α]; Diaclone), in-house ELISA (IFN-α) developed using commercially available antibodies (from BD Pharmingen or PBL, respectively), or by the Luminex technology using a Cytokine 25-Plex for Luminex™ (BioSource).
Reporter assay
HEK-293 cells containing an nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NFκB)-Luciferase reporter construct and expressing human TLR7, TLR8, or TLR9, or without TLR expression were used as described before (Jurk et al., 2002, 2006b; Gorden et al., 2005). Cells were plated on 96-well plates at 1.5×104/well and allowed to attach overnight. The cells were subsequently incubated for 16 h with the indicated amount of ORNs, CpG ODN, or ORN/CpG ODN chimera complexed to DOTAP or DOTAP alone and then tested for luciferase expression. Each data point was done in duplicate.
Polyacrylamide gel electrophoresis gel
About 3.75 mL 40% polyacrylamide, 1 mL 10×Tris-Borat-EDTA-buffer, 5.75 mL DNA/RNAse-free water, 100 μL 10% ammonium persulfate, and 10 μL Tetra-methyl-ethylene-diamine (all Sigma-Aldrich) were mixed and filled into a gel chamber. After 15 min of hardening, 10 μL of 1 μg ORN was mixed with 2 μL loading buffer and added to the gel pockets. The gel was incubated at 100 V and 0.2 A for 1–2 h and finally stained with StainsAll (Sigma-Aldrich).
Murine in vitro assays
sv129 mice (6–8 weeks of age) were used for all experiments and purchased from Charles River Canada and housed in microisolators at the animal care facility of Pfizer Vaccine. ORNs formulated with or without DOTAP were administered iv to sv129 mice (n=5 per group) and after 3 h postinjection animals were bled and interferon gamma-induced protein 10 (IP-10) levels in plasma were measured by ELISA (PharMingen).
Murine splenic DCs were derived from sv129 mice using CD11c+ immunomagnetic bead positive selection according to manufacturer's recommendations (Miltenyi Biotech). Murine cells were added to 96-well round-bottomed plates with or without the addition of reagents as indicated. Culture SNs were collected at 20 h and cytokine amounts were assessed using a commercially available ELISA kit (BD Pharmingen) or an in-house murine IFN-α ELISA developed using commercially available antibodies (from PBL).
Mice were housed in microisolator cages in the Animal Care Facility at the Coley Pharmaceutical Group, Canada. All studies were approved by the Animal Care Committee of Coley Canada and were conducted in accordance with the procedures of the Animal Care Facility at Coley Canada under the guidance of the Canadian Council on Animal Care.
Results
3′ Poly G tails are sufficient to induce an immune response by unformulated ORNs
Previous studies demonstrated sequence-dependent immune activation of TLR7 and TLR8 by DOTAP delivered GU-rich ORNs (Heil et al., 2004; Hornung et al., 2005; Forsbach et al., 2007, 2008). DOTAP has been described by several publications as uptake enhancer and for ORN protection against exo- and endoribonucleases. Due to their cell type–specific expression, TLR7 stimulation triggers preferentially IFN-α production, whereas TLR8 stimulation triggers other, for example, proinflammatory cytokines such as TNF-α and IFN-γ (Gorski et al., 2006; Forsbach et al., 2008).
3′ Poly G tails in CpG ODN TLR9 agonists have been shown to self-associate via Hoogsteen base pairing to form parallel quadruplex structures called G-tetrads (Costa et al., 2004; Puig et al., 2006). For the so-called A-Class ODN, the formation of multimers is necessary for localization in early endosomes, and for higher stability, resulting in pDC-mediated type I IFN immune response (Anderson et al., 1996; Krug et al., 2001b; Gursel et al., 2002; Vollmer et al., 2004b; Kerkmann et al., 2005), stronger than observed for most other CpG classes. This structure and stability information was used in this study to design ORNs, which may not require endosomal delivery systems. A poly rG (R-2176) or poly dG (R-1935) tail was added 3′ to the GU-rich TLR7/8 activating ORN R-0006 (Krieg et al., 1995) with the aim to stabilize the unformulated ORNs against 3′ exonucleases, to enhance uptake, and to induce strong type I IFN. Figure 1 shows the in vitro human PBMC immune response in the absence or presence of the delivery system DOTAP. Whereas DOTAP formulation of ORN R-1935 and R-2176 resulted in a 1 log decrease in potency, and in about 1/3 loss of maximum activity compared with R-0006 for IFN-α and IFN-γ, only those unformulated ORNs containing a poly G tail induced type I IFN secretion upon human PBMC activation. No difference was observed for IFN-α release in the absence of DOTAP between an RNA or DNA poly G tail. Comparison of DOTAP formulated and unformulated R-1935 and R-2176 showed strongly decreased type I IFN for unformulated R-1935 and R-2176. However, the observed immune response is still significant. Here, we protect only the 3′ but not the 5′ end or degradation by endonucleases. Therefore, those unformulated ORNs contain lower stability which leads to lower immune response compared to DOTAP-formulated ones.

Human PBMCs were activated by formulated and unformulated ORNs containing 3′ poly G tails. Poly G tails result in an immune response stimulated by unformulated ORNs. Human PBMCs (3 individual blood donors) were stimulated by the indicated concentrations of ORNs complexed to 25 μg/mL DOTAP or in the absence of DOTAP (-DOTAP). Medium and LPS (100 ng/mL) were used as assay controls. After 24 h, supernatants were harvested and IFN-α
TLR8-mediated immune effects could not be observed upon culture of human immune cells with poly G ORNs, indicating that TLR7 but not TLR8 is involved in the observed immune effects. These data suggest that adding G-tetrads to form higher-ordered structures in immune stimulatory ORNs offers the possibility to use such single-stranded RNAs without delivery systems more potently for TLR7 immune activation.
Chemical modifications like 2′-O,4′-C-methylene bridged nucleotides (LNA), 2′-O-methyl (m), or 3′-O-methyl (3m) are known to stabilize toward nuclease digestion and might be alternatives to desoxy- or ribo-G tails (Crinelli et al., 2002; KURRECK, 2003). Furthermore, single nucleosides such as 8-oxo-2′-deoxy-guanosine (dO) and 8-bromo-2′-deoxy-guanosine (dBG) were previously shown to stimulate TLR7-mediated immune responses, and, therefore, may increase activity of ORNs containing 3′ poly G overhangs (Smee et al., 1989; Sharma et al., 1992a; Pope et al., 1995; Lee et al., 2003; Gros et al., 2007). ORN R-1935 with poly rG was modified at the 3′ guanosine by introducing a 3mG or dT, by adding an LNA or 2′-O-methyl G tail (LNA or m), or by modifying the first guanosine of the poly G tail (dO or dBG; Fig. 2). All ORNs when formulated stimulated IFN-α cytokine production with minor differences in maximum activities (Fig. 2B). The greatest impact with a 50% cytokine reduction compared with the parent ORN R-0006 (first ORN in Fig. 2) and poly rG ORN R-1935 (second ORN in Fig. 2) was observed for the modification of all 3′ guanosines by 2′O-methyl and LNA or the modification of the 5′ guanosine of the poly G tail by either dO or dBG. A negative influence of 2′O-methyl modifications on immune stimulatory single- and double-stranded RNA has been described previously (Robbins et al., 2007; Sioud et al., 2007; Tluk et al., 2009), as well as of LNA modifications on the immune response for TLR9 nucleic acid agonists that, therefore, can be expanded to include TLR7/8 nucleic acid agonists (Vollmer et al., 2004a). However, the results with dO or dBG modifications are in contrast to the literature (Smee et al., 1989; Sharma et al., 1992a; Pope et al., 1995; Lee et al., 2003; Gros et al., 2007), which may be due to the incorporation of these modifications in the 3′ poly G tail. A similar alteration of the immune stimulatory activity was observed for unformulated poly G tail ORNs (Fig. 2A). The addition of a 3′-O-methyl-guanosine at the 3′ end of the poly G tail did not result in a substantial effect on the type I IFN immune response of unformulated ORNs; however, the addition of a 3′ dT resulted in a somewhat stronger immune effect at the concentration tested (Fig. 2A). This may be due to the differences in G-tetrad formation (Fig. 3 and data not shown). Interestingly, unformulated ORNs with a 3′ poly dG tail but phosphodiester linkages did not lead to type I IFN production in contrast to ORNs with phosphorothioate linkages, except for the addition of a 3′-O-methyl-guanosine at the 3′-terminus. This may be due to stronger 3′ exonuclease resistance of phosphorothioate than phosphodiester nucleotide linkages (Furdon et al., 1989; Gilar et al., 1998). Chemical modification of the phosphodiester poly G tail by a 3′-O-methyl-rG retained type I IFN production. A single 3′-O-methyl-guanosine has the ability to protect against 3′-ribo-exonucleases (Kumar and Takaku, 1999).
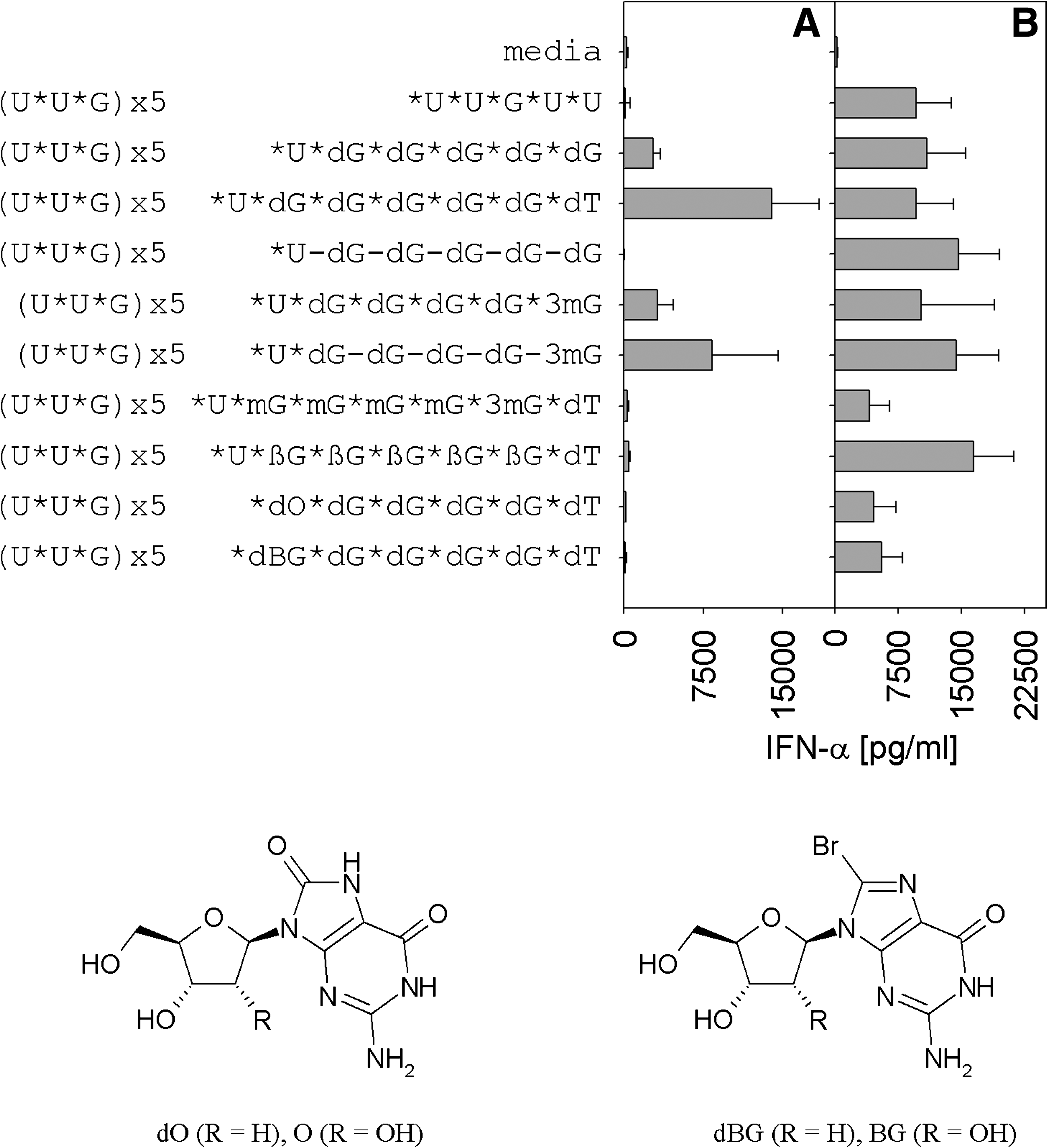
Human PBMCs were activated by formulated and unformulated ORNs containing chemical modifications in the 3′ poly G tails. Chemical modifications in the poly G tail do not further improve immune effects, but rather decrease cytokine secretion. Human PBMCs (3 individual blood donors) were stimulated by 1.3 μM ORN alone
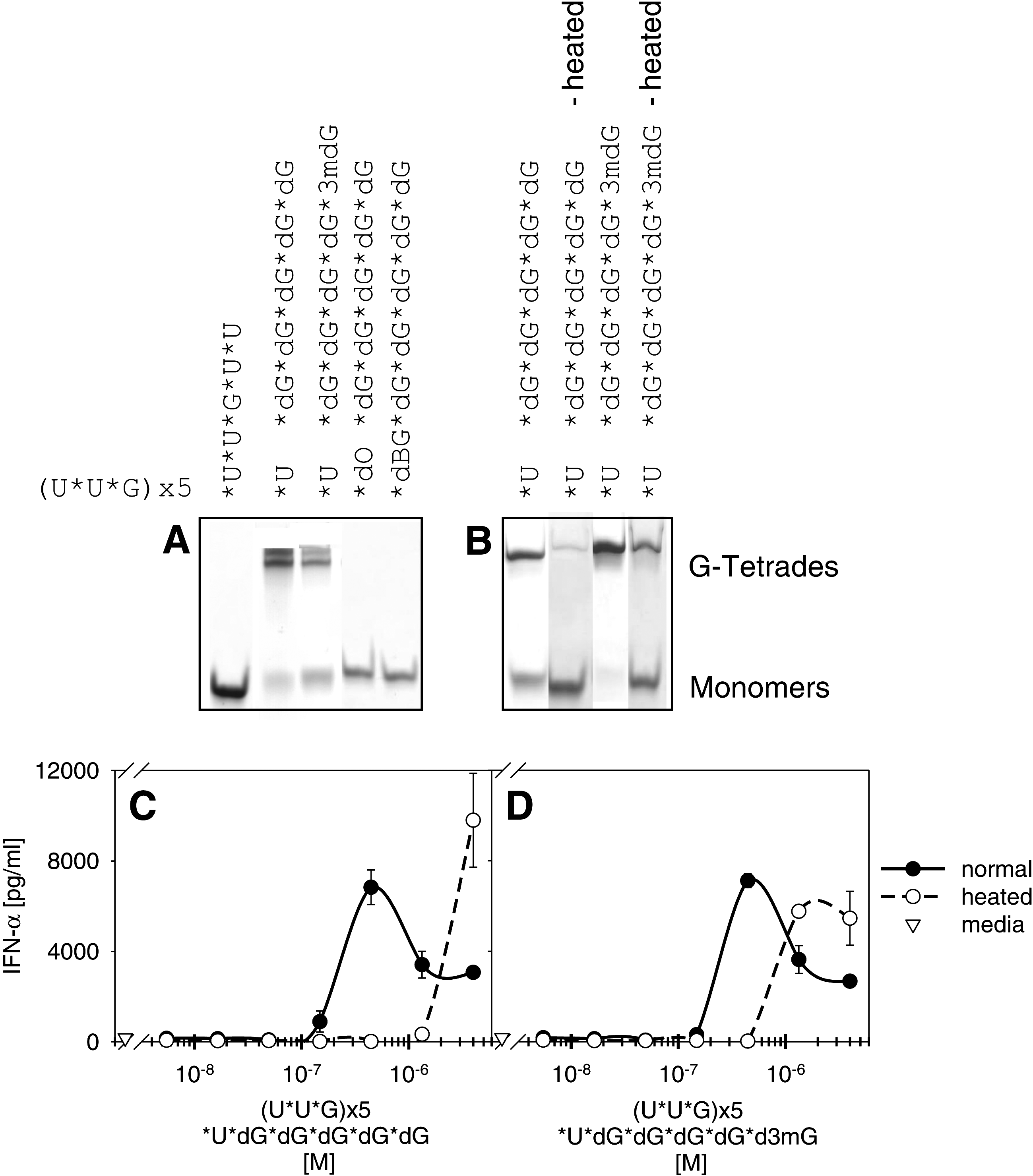
G-tetrad measurement by PAGE gel combined with human PBMC activation of 3′ poly G tail containing ORNs. Type I interferon production induced by unformulated ORNs depends on G-tetrad formation.
As shown in Fig. 3 the type I IFN induction of unformulated poly G tail ORNs correlates with the extent of ORN G-tetrad formation. Only those 3′ poly G–modified ORNs forming G-tetrads are capable to induce type I IFN without DOTAP formulation (compare Figs. 2A and 3A). For example, modification of the poly G tails with a 5′ dO or dBG suppresses IFN-α cytokine production. Indeed, by measuring tetrad formation, no ORN aggregation is observed for these modified poly G ORNs. Heating of poly G tail ORNs dissociates the G-tetrads as demonstrated in Fig. 3B, and substantially decreases the IFN-α cytokine production (Fig. 3C+D). However, although the results using poly G tail ORNs look promising, a clinical development of such ORNs most probably will have certain limitations due to the formation of high-ordered structures that are difficult to resolve.
In addition to poly G tails, cholesterol, but not other chemical 3′ modifications, improves TLR7 immune responses
Figure 4 summarizes the type I IFN and TNF-α immune response induced by R-1219—a short GU-rich ORNs—and derivates thereof containing different 3′ additions like cholesterol (chol), triethyleneglycol (teg), acridine (acr), 6-carboxyfluorescein (fam), biotin (biot), 1,2-di-O-hexadecyl-rac-glycerol (hex), or a poly rG RNA tail. The 3′ additions were added to GU-rich ORNs with the aim to test additional modifications potentially resulting in protection against 3′ ribo-exonucleases and ORN usage without endosomal delivery systems.

Human PBMCs in vitro and mice in vitro and in vivo studies of ORNs with 3′ chemical modifications. 3′ Modifications other than poly G or cholesterol do not improve the immune response stimulated by unformulated ORNs.
Immune activation was tested for unformulated ORNs (Fig. 3A, C) or ORNs formulated with DOTAP (Fig. 3B, D). Beside the poly G tail, also a 3′ cholesterol results in TLR7-mediated type I IFN release by unformulated ORNs. Although immune effects are observed, these were lower than for encapsulated ORNs similar to Fig. 1 (Fig. 4A–G). Interestingly, only unformulated 6-FAM and acridine 3′ modifications resulted in TLR8-mediated TNF-α cytokine release upon human immune cell activation without affecting TLR7-dependent IFN-α production. Upon encapsulation, poly G tail, 1,2-di-O-hexadecyl-rac-glycerol (hex), and to some extent cholesterol modifications increased the type I IFN response, but only the cholesterol modification increased also the TNF-α production compared with R-1219. Similar effects as observed for human immune cells were obtained by using murine immune cells (Fig. 4F, G). Both poly G and cholesterol modifications resulted in immune stimulatory effects by unformulated ORNs. However, in contrast to human immune cells, the cholesterol modification appears to decrease murine immune effects upon encapsulation (Fig. 4G). However, this may be due to the experimental conditions used as both these modified ORNs resulted in substantial IP-10 production upon iv administration (Fig. 4F) in the absence of a delivery system, and encapsulation into DOTAP further increased chemokine secretion. In summary, we describe different chemical ORN modifications activating the immune system in vitro and in vivo in the absence of delivery reagents: ORNs with poly G tails or with 3′ cholesterol stimulating TLR7, and ORNs with 3′ 6-FAM or 3′-acridine stimulating TLR8.
Further detailed analysis of the effects of poly G tail or cholesterol modifications revealed for cholesterol a length dependency for TLR7-mediated type I IFN responses (Fig. 5A). Shorter GU-rich ORNs with a 3′ cholesterol modification resulted in higher IFN-α release (Fig. 5A) and stronger potency (data not shown) than longer such ORNs. No effect of different lengths was observed for poly G tail ORNs or TLR8-mediated TNF-α production (Fig. 4A, B). Cholesterol-modified ORNs may represent an alternative approach to use ORNs without delivery system, although cholesterol ORNs can also form high-ordered structures (data not shown).

Human PBMCs were activated by unformulated ORNs containing 3′ poly G tails or 3′ cholesterol. 3′ Cholesterol modified, but not poly G modified, ORNs show length dependency for type I interferon secretion in the absence of a delivery system. Human PBMCs (3 individual blood donors) were stimulated by 1.3 μM ORNs complexed to 8.3 μg/mL DOTAP. Medium or ORN R-0006 ((U*U*G)x 6 *UU) with and without DOTAP were used as assay controls. After 24 h, supernatants were harvested and IFN-α
Specific chemical modifications of guanosine or uridine nucleotides in GU-rich ORNs stimulate type I IFN immune responses via TLR9
We tested in the second part of this study combined TLR7, TLR8, and/or TLR9 immune responses. We tested several chemical modifications to combine and/or convert the ORN-mediated TLR7 and TLR8 immune response to TLR9.
Single-guanosine ribonucleoside analogues like 7-deaza-, 8-bromo-, or 8-oxo-guanosine were described to activate the immune system via TLR7 (Lee et al., 2003) or to enhance type I IFN production in case of central incorporation in single-stranded RNA (Pope et al., 1995; Busconi et al., 2006). Beside these modifications we used the chemical modifications 7-deaza-adenosine, universal purine, pyrimidine ribonucleotides, inosine, and 5-fluoro-modified uridine to substitute for U or G, and tested their effect on TLR7 and TLR8 activation. Immune activation was tested upon culturing human PBMCs (Figs. 6 and 7) or stable TLR7-, TLR8-, and TLR9-transfected HEK-293 cells (Fig. 7 and data not shown). The guanosine analogues 7-deaza-guanosine, 7-deaza-adenosine, inosine, and universal purine ribonucleotides (2-amino-purine-ribonucleotide and purine-ribonucleotide) decrease potency and maximal activity of type I IFN production compared with the parent ORN R-2050 with unmodified guanosines. Similar results were observed for uridine exchanges by either 5-fluoro-ribo-uridine or a universal pyridine nucleotide (pyridine-2-on-ribonucleotide). However, TLR8-mediated IFN-γ release upon human immune cell activation was affected to a lower extent compared with the type I IFN production (Fig. 6B).
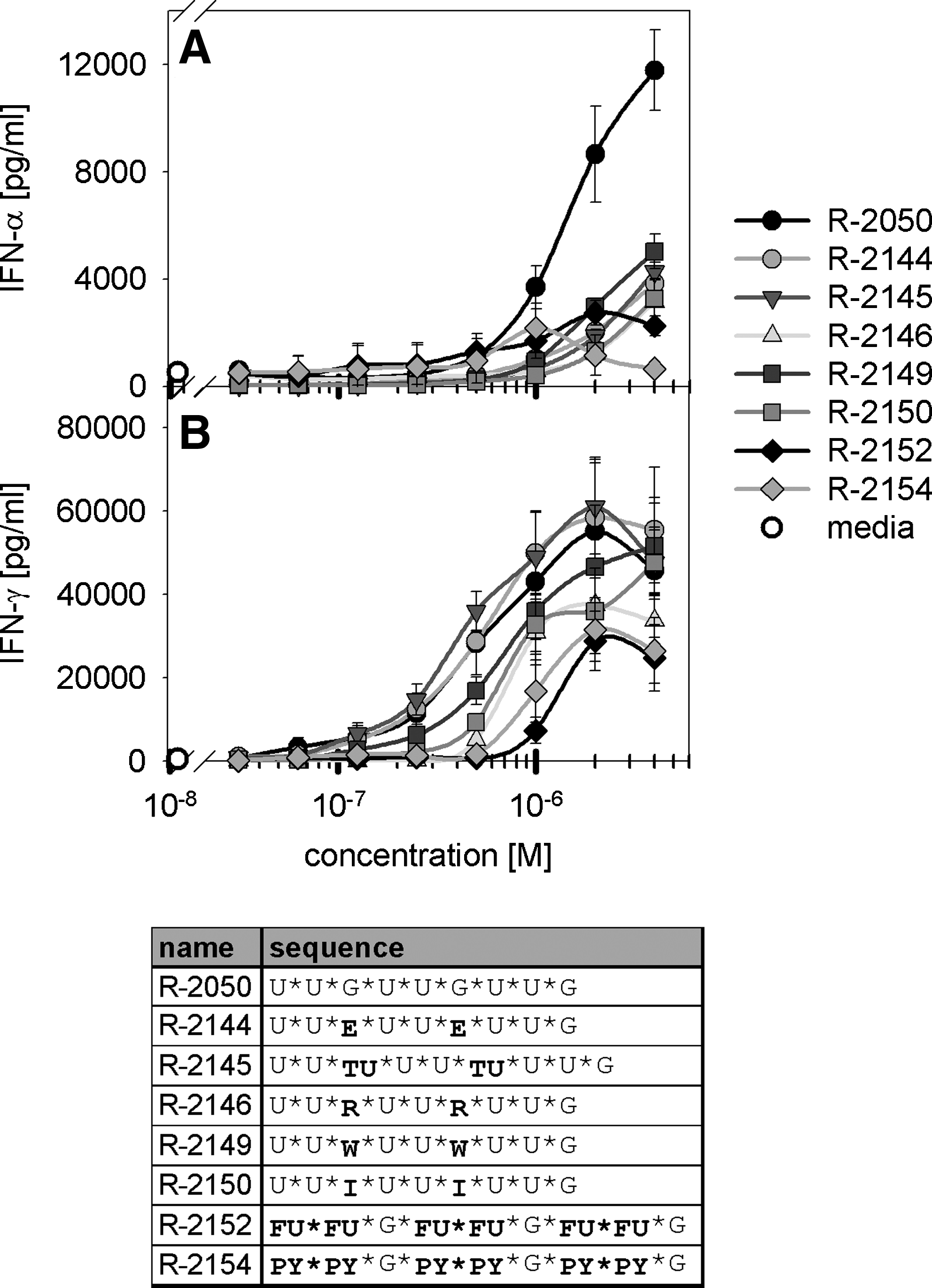
Human PBMCs were activated by chemically modified ORNs. Specific chemical modifications of G or U decrease TLR7-mediated, but not TLR8-mediated, immune effects. Human PBMCs (3 individual blood donors) were stimulated by the indicated ORN concentrations complexed to 25 μg/mL DOTAP (1/3 dilution). Medium was used as assay control. After 24 h, supernatants were harvested and IFN-α
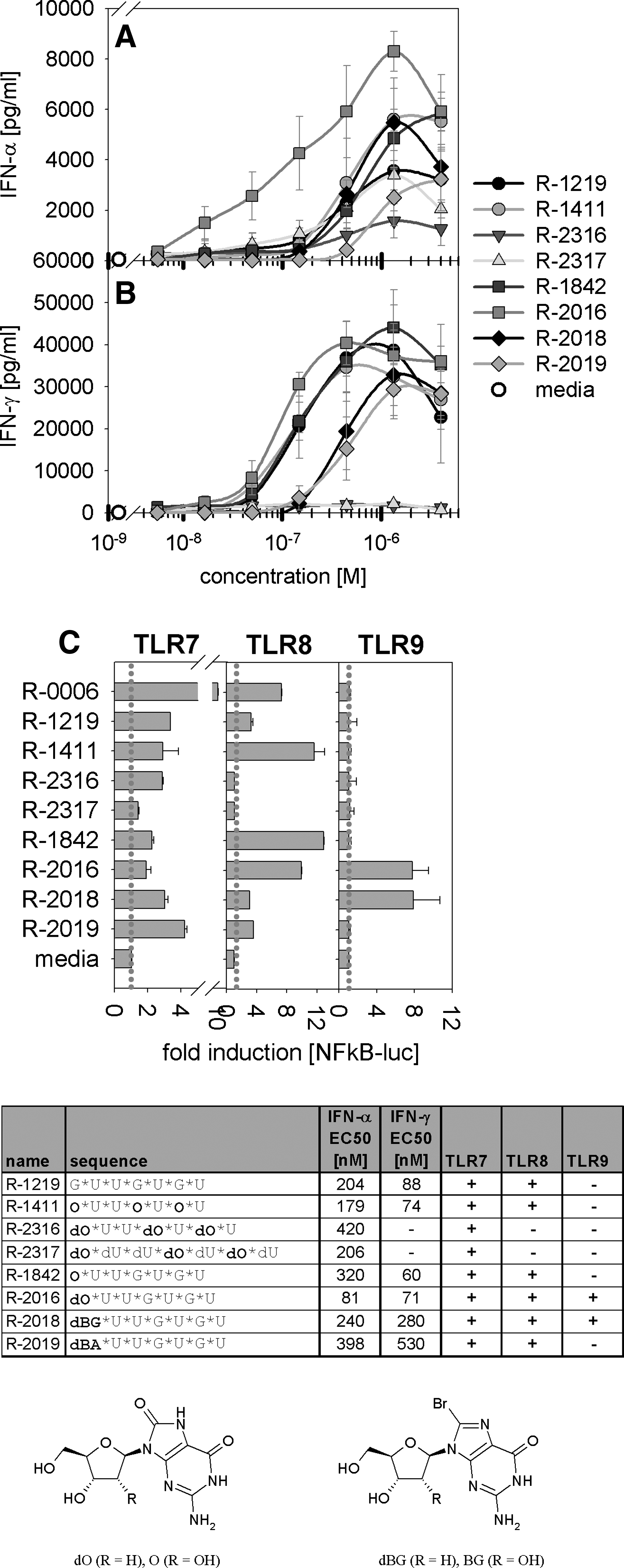
Human PBMCs and stable transfected TLR7, TLR8, or TLR9 HEK-293 cells were activated by ORNs with specific 5′ chemical modifications. Specific 5′ chemical modifications result in TLR9-dependent immune activation. Human PBMCs (3 individual blood donors) were stimulated by the indicated ORN concentrations complexed to 25 μg/mL DOTAP (1/3 dilution). Media were used as assay control. After 24 h, supernatants were harvested and IFN-α
These data suggest that nucleotide analogues or universal purine/pyridine nucleotides for either all guanosines or all uridines negatively affect TLR7-dependent immune effects by single-stranded GU-rich ORNs. However, single 5′ 8-oxo-2′-deoxy-guanosine modifications (as in ORN R-2016) resulted in a 100% increase of maximal activity and a 2.5-fold increase of potency of the type I IFN immune response compared with the unmodified GU-rich ORN R-1219 (Fig. 7). Beside R-2016, also ORN R-2018 (with a single 5′ 8-bromo-2′-deoxy-guanosine), R-1411 (exchange of all G by 8-oxo-guanosine), and R-1842 (single 5′ 8-oxo-guanosine) resulted in an increase in maximal IFN-α activity compared with parent ORN R-1219, whereas R-2316 (exchange of all G by 8-oxo-2′-deoxy-guanosine) and R-2019 (single 5′ 8-bromo-2′-deoxy-guanosine) showed decreased responses (Fig. 7A). TLR8-mediated IFN-γ production upon activation of human immune cells was not altered when compared with the parent ORN R-1219 (Fig. 7B). Further experiments (data not shown) demonstrated that for GU-rich ORNs only a modification of the 5′ guanosine, but not of the central or 3′ guanosine, with 8-oxo-2′-deoxy-guanosine results in an increased IFN-α response. Further investigation of the effect of such modifications on the TLR8 response revealed that a 5′ 8-bromo-2′-deoxy-guanosine or -adenosine modification as well as modifying all G by 8-oxo-2′-deoxy-guanosine results in decreased TLR8-mediated effects (using IFN-γ as a read-out for TLR8) compared with unmodified ORN R-1219 (Fig. 7C).
Stable transfected cell lines expressing TLR7, TLR8, or TLR9 were used to investigate whether the enhanced type I IFN production observed with R-2016 (8-oxo-2′-deoxy-guanosine modification of the first 5′ guanosine) is due to signaling other than TLR7 (Fig. 7C). Surprisingly, both ORN R-2016 and R-2018 (8-bromo-2′-deoxy-guanosine modification of the first 5′ guanosine) induced TLR9-mediated NFκB signaling, demonstrating that these 5′ modifications result in triggering of TLR9 responses. These data show that TLR7/8-mediated immune responses induced by GU-rich ORNs can be expanded to include TLR9 by a simple chemical modification of the first 5′ guanosine by either 8-oxo-2′-deoxy-guanosine or 8-bromo-2′-deoxy-guanosine. No TLR9-dependent signaling or an increased IFN-α response could be observed for 8-bromo-2′-deoxy-adenosine compared with 8-bromo-2′-deoxy-guanosine, suggesting that the induction of TLR9 activation is limited to 5′ guanosine analogues.
Taken together, 5′ modification of short ORNs with 8-oxo-guanosine or 8-bromo-2′-deoxy-guanosine results in combined activation of TLR7, TLR8, and TLR9. Similar data have been observed with longer ORNs (data not shown). Due to the decreased TLR8-mediated immune response of the 5′ 8-bromo-2′-deoxy-guanosine–modified ORNs, it may even be possible to further develop ORNs that mainly drive TLR7 and TLR9 responses without strong production of proinflammatory cytokines via TLR8. In addition, 5′-modified ORNs redefine the PBMC definition of cytokine selectivity with type I IFN and TNF-α/IFN-γ for TLR7 and TLR8. Here, type I IFN immune response may, beside TLR7, also indicate for TLR9 and real TLR activation data are needed.
RNA-DNA chimera stimulates the innate immune system via TLR8 and TLR9, but lacks TLR7 activation
Beside the issue of chemical modifications, we combined classical CpG-ODN and GU-ORN motifs and tested whether these motifs would activate TLR7, TLR8, and TLR9. The typical C-Class CpG ODN 2395, a DNA TLR9 agonist, shows TLR9-dependent type I IFN secretion, but very limited IFN-γ or TNF-α cytokine production upon human PBMC activation (Vollmer et al., 2004b). To generate RNA-DNA chimeras potentially stimulating CpG-mediated TLR9 and RNA-mediated TLR7/8 immune effects, we displaced the palindromic tail in ODN 2395 by GU-rich (TLR7/8 stimulatory) or GA-rich (nonstimulatory control) single-stranded RNA sequences and added 4 desoxy thymidines at the 3′ end (to protect the 3′ end of the DNA-RNA chimer). The ORN R-2322 and R-2326 consist of a 5′ CpG motif and either a GU-rich (R-2322) or GA-rich (R-2326) RNA sequence, whereas R-2323 and R-2327 are GpC controls that should not stimulate TLR9 signaling (Fig. 8). The induction of immune responses was investigated by using TLR7, TLR8, or TLR9 stable transfected HEK-293 cells (Fig. 8A), or human immune cells (Fig. 8B, C). Similar to previously published data (Vollmer et al., 2004b; Forsbach et al., 2008), the TLR7/8 agonist ORN R-0006 activated TLR7 and TLR8 and induced type I IFN and IFN-γ secretion, and CpG ODN 2395 activated TLR9 and induced type I IFN cytokine production (Fig. 8A–C). Chimeric oligonucleotides did not show stronger or additive TLR7 and TLR9 effects for type I IFN responses (Fig. 8A). Chimera R-2326 (with a GA-rich RNA sequence) does not contain a stimulatory TLR7 or TLR8 motif, but this oligonucleotide induced IFN-α cytokine production comparable to CpG ODN 2395, whereas chimera R-2323 (with a GpC DNA sequence) does not contain a stimulatory TLR9 motif and, although having a GU-rich sequence, failed to induce type I IFN secretion. Therefore, due to the lack of IFN-α stimulation in the absence of a CpG motif, it can be concluded that the IFN-α release by chimeric RNA-DNA oligonucleotides is dependent on TLR9 but not TLR7. Indeed, although containing a GU-rich RNA sequence, neither ORN R-2322 nor R-2323 stimulated TLR7-mediated NFκB signaling in HEK-293 cells (Fig. 8A). Previous data demonstrated that T-rich phosphorothioate DNA oligonucleotides are inhibitory to TLR7 activation (Jurk et al., 2006b). Experiments with stable TLR7-, TLR8-, or TLR9-transfected HEK-293 cells demonstrated that none of the tested chimera was able to activate TLR7, but stimulated CpG-dependent TLR9 signaling (Fig. 8A). Further experiments aimed at investigating this inhibition in more detail. As shown in Fig. 9, the type I IFN immune response by a single-stranded TLR7/8 RNA agonist of the same length and composition as in the chimeric molecules, ORN R-2204, is blocked by a GpC ODN (R-24068; again of the same length and sequence as in the chimeric molecule), but not by such a CpG ODN (ODN 5435). Due to the CpG dinucleotides present in ODN 5435, an inhibitory effect on the ORNs was not expected to be observed as the CpG ODN itself induces IFN-α secretion.

Human PBMCs and stable transfected TLR7, TLR8, or TLR9 HEK-293 cells were activated by RNA-DNA oligonucleotide chimera. RNA-DNA oligonucleotide chimera activates the immune system via TLR8 and TLR9.

Human PBMC immune response was analyzed upon ORN and phosphorothioate DNA activation. TLR7/8-mediated cytokine production by ORNs is inhibited by phosphorothioate DNA. Human PBMCs (3 individual blood donors) were stimulated with 1 μM of ORN R-2204 complexed to 12.5 μg/mL DOTAP or 1 μM of ORN R-2204 complexed to 12.5 μg/mL DOTAP and the indicated concentrations of CpG ODN. After 24 h, supernatants were harvested and IFN-α
Although not stimulating TLR7-mediated immune responses, the GU-containing chimera still activate TLR8 in a sequence-specific manner as shown for R-2322 and R-2323 (Fig. 8A, C). Chimeric molecules with a GA-rich RNA sequence (R-2326 and R-2327) failed to activate TLR8-mediated effects (Fig. 8A, C). In addition, no blockage of TLR8 signaling by single-stranded DNA could be observed for these chimeric molecules (Fig. 8) or by adding short phosphorothioate CpG or GpC ODN (5435 or 24068, respectively) to a single-stranded ORN, R-2204 (Fig. 9B). Similar effects were observed in in vivo experiments (Fig. 8D and data not shown).
To further analyze immune effects stimulated by chimeric RNA-DNA versus RNA or DNA oligonucleotides, activation of human immune cells by a B-Class CpG ODN TLR9 agonist 10103, a TLR7/8 agonist ORN R-1219, and chimeric oligonucleotides [R-1626 (CpG ODN with GA-rich RNA), R-1228 (GpC ODN with GU-rich RNA), and R-1226 (CpG ODN with GU-rich RNA)] was analyzed in a Luminex assay to measure several different cytokine and chemokines (Fig. 10). Cytokine and chemokine secretion induced by the RNA-DNA chimera indeed can be separated in DNA-, RNA-, and DNA/RNA-dependent effects. Comparison of chimera R-1226 and R-1228 demonstrated that only type I IFN is TLR9 mediated and CpG dependent. As shown in Fig. 9, GpC-ODN/GU-ORN chimera R-1228 failed to induce type I IFN whereas CpG-ODN/GU-ORN chimera R-1226 induced TLR9-mediated IFN-α release. IL-1Rα, IL-1β, IL-7, IL-8, IL-12p40, IL-13, IFN-γ, granulocyte macrophage colony-stimulating factor (GM-CSF), macrophage inflammatory protein (MIP-1)α, MIP1-β, MIG, TNF-α, and Eotaxin induction is TLR7/8 mediated and RNA dependent, because CpG-ODN/GA-ORN chimera R-1626 failed to induce these cytokines and chemokines compared with CpG-ODN/GU-ORN chimera R-1226. In contrast, RNA- and DNA-dependent effects were observed for IL-2R, IL-6, IL-10, IP-10, monocyte chemoattractant protein-1 (MCP-1), and Rantes for all tested chimera, as GpC- and GA-rich sequences each reduced the combined effects.

Human PBMCs were activated by RNA-DNA chimera. Cytokine and chemokine responses of RNA-DNA chimera are DNA- and/or RNA-dependent effects. Human PBMCs (3 individual blood donors) were stimulated with 0.12 μM of ORN, CpG ODN, or RNA-DNA chimera complexed to DOTAP (1.5 μg/mL). After 24 h, supernatants were harvested and cytokines of each individual donor were separately measured by Luminex. Data shown are mean (±SEM) maximal activities (pg/mL) of 3 donors. DOTAP and medium were used as assay controls. IL-2, IL-4, IL-5, IL-15, and IL-17 did not show any detectable cytokines. Sequences are listed. TNF, tumor necrosis factor; MCP-1, monocyte chemoattractant protein-1; IP-10, interferon gamma-induced protein 10; MIP-1, macrophage inflammatory protein; GM-CSF, granulocyte macrophage colony-stimulating factor.
In summary, RNA-DNA chimeric oligonucleotides are combined TLR9 and TLR8 agonists, and demonstrate specific effects for the CpG ODN and GU-rich RNA sequences. Together with the above results we identified oligonucleotides with the capability to stimulate either TLR7/8, TLR7/8 and 9, TLR7 and TLR9, or TLR8 and TLR9. This expands the described classes of TLR7/8/9 oligonucleotide agonists to molecules stimulating all combinations of these receptors that could be used for specific disease indications.
Discussion
TLR7, TLR8, and TLR9 belong to a subfamily of TLRs and evolved as nearest neighbors. TLR7 and TLR8 were shown to build 1 subgroup in this family (Chuang and Ulevitch, 2000; Du et al., 2000). Specific recognition of single-stranded RNA via human TLR7 and TLR8 has been already described (Heil et al., 2004; Hornung et al., 2005; SIOUD, 2005; Vollmer et al., 2005). Differentiation between TLR7, TLR7/8, and TLR8 immune activation was published for small molecules (Gorden et al., 2005; Gorski et al., 2006) and recently for single-stranded RNAs or synthetic ORNs containing specific RNA sequence motifs (Forsbach et al., 2007, 2008; Jurk et al., 2011). Nonmethylated, CpG-containing DNA of bacterial or viral origin or synthetic CpG ODN has been identified as TLR9 ligands.
Human TLR7, TLR8, and TLR9 activate divergent cell types due to their cell-specific expression. TLR7 agonists directly activate human pDCs to produce type I IFN and related genes (like MIP1-a, MIP1-b, Rantes or IP-10); TLR8 agonists directly activate mDCs, monocytes, and NK-cells to produce proinflammatory cytokines and chemokines like TNF-α or IFN-γ; and TLR9 agonists result in human pDC and B cell activation characterized by the induction of IFN-α and B cell–related cytokines like IL-10 and IL-6 (Gibson et al., 2002; Heil et al., 2004; Vollmer et al., 2004b; Hornung et al., 2005; Gorski et al., 2006; Jurk et al., 2006a). This diversity and target-specific cell selectivity is a useful tool to analyze potential TLR7, TLR8, or TLR9 ligands and combinations thereof. In combination with recombinant cells expressing TLR7, TLR8, or TLR9, this enables the identification and characterization of single, dual, or triple TLR ligands. One of our goals was the identification of ORNs to be efficiently used without the addition of delivery systems. Single-stranded RNA molecules undergo stronger endo- and exonuclease digestion compared with single-stranded DNA molecules (Cummins et al., 1996; Leeds et al., 1996; Gilar et al., 1997) and require therefore specific delivery systems, not only to protect these more labile molecules, but also to transport them to their target cells and TLR-containing intracellular compartments. In this study, we developed chemically modified ORNs that do not require endosomal delivery systems such as DOTAP. This opens the possibility of lower toxicity and side effects of ORNs during clinical development. By introducing specific 3′ modifications or chemical modifications of guanosine or uracil, we identified ORNs activating TLR7 or TLR8 without a delivery system. The most promising of such modifications were shown to be either cholesterol or 5 guanosines at the ORN 3′ end, although these stimulated immune effects at lower potency and efficacy compared with encapsulated ORNs. This may be due because the unformulated ORNs can be still strongly affected by endo- or exonucleases than the encapsulated ones. As described previously, a stretch of 3′ guanosines self-associates via Hoogsteen base pairing to form parallel quadruplex structures called G-tetrads (Costa et al., 2004; Puig et al., 2006). This G-tetrad formation is most likely responsible for increased ORN stability and type I IFN immune response of unformulated ORNs, and can be compared with the so-called A-Class of CpG ODN TLR9 agonists (Hartmann et al., 2003). ORN aggregation in the context of a 3′ cholesterol modification was observed as well (data not shown), and such cholesterol-modified ORNs also stimulated immune effects in the absence of a specific delivery system such as DOTAP. Thus, similar to DNA TLR9 agonists, the formation of stable higher-ordered structures by adding 3′ cholesterol or poly G results in TLR7-mediated type I IFN and other immune response of such unformulated molecules. Avoiding delivery systems for ORNs may have greater advantages for a potential clinical development, as it reduces the required dose to induce specific immune effects compared with unmodified unformulated ORNs, although the synthesis and quality control of substances forming secondary or tertiary structures have specific challenges.
Chemical modifications of all uridines or guanosines in GU-rich ORNs could not overcome the need for a delivery system, although such modifications are expected to increase the stability of an ORN (Smee et al., 1989, 1995; Sharma et al., 1992a, 1992b; Pope et al., 1995; KURRECK, 2003; Lee et al., 2003). In contrast, such modifications at all uridines or guanosines even decrease TLR7-mediated and TLR8-mediated immune effects.
Interestingly, we showed that chemical modifications expanded the immune response of TLR7/8 activating ORNs to TLR9. Combined TLR7, TLR8, and TLR9 immune responses open several new possibilities and treatments in clinical development. We explored in detail that chemical modification of only the first 5′ guanosine by 8-oxo-2′-deoxy-guanosine, 8-oxo-guanosine, or 8-bromo-2′-deoxy-guanosine resulted in activation not only of TLR7/8, but also stimulated TLR9-mediated effects. Such modified ORNs further induce additive effects for type I IFN production. In contrast to the dO or 8-oxo-guanosine modification that retained the capability to induce TLR8-dependent effects, 8-bromo-2′-deoxy-guanosine modification of the first 5′ guanosine resulted in a decreased TLR8 immune response. This implies that the choice of the 5′ modification defines which TLR (TLR7, TLR8, and/or TLR9) can be combined. Such ORNs open the possibility to develop triple or dual ligands for TLR7, TLR8, and/or TLR9. In addition, diversity and target-specific cell selectivity for 5′-modified ORNs requires beside human PBMCs also the combination with recombinant cells expressing TLR7, TLR8, or TLR9 to conclude which TLR has been activated.
Oxidative damage to cellular components has been proposed to play an important role in the development of a number of pathological processes including cancer (Loft et al., 1995; Loft and Poulsen, 1996). In addition, an accumulation of damages in nuclear and mitochondrial DNA often appears in aging and inflammatory processes including atherosclerosis, diabetes, and neurodegenerative diseases (Stevnsner et al., 2002; Thorslund et al., 2002). 8-oxo-2′-deoxy-guanosine and 8-oxo-guanosine are one of the most occurring mutagenic DNA and RNA base modifications in these diseases and used as disease marker (Weimann et al., 2002; Banerjee et al., 2005; Haghdoost et al., 2005, 2006). The repair enzyme oxoguanine DNA glycosylase is one important enzyme involved in base excision repair processes to eliminate oxidative damage from mammalian DNA. However, TLR7, TLR8, and TLR9 activation may be an alternative response to oxidative damage of the human system in protection against cancer or other age-related diseases. Furthermore, 8-oxo-2′-deoxy-guanosine– and 8-oxo-guanosine–modified ORNs representing specific TLR7/8/9 agonists and inducing a broad spectrum of cytokines and chemokines appear as potential candidates for treatment of cancer and inflammatory diseases.
Similar results to combine TLR9 and TLR7/8 effects were achieved by using chimeric molecules consisting of a CpG ODN sequence as TLR9 agonist, and a GU-rich single-stranded ORN as TLR7/8 agonist. Although these chimeric oligonucleotides were expected to stimulate all three TLRs, the effects induced by these molecules are limited to TLR8 and TLR9 activation. The TLR7-mediated human type I IFN immune response is suppressed by a sequence-independent but phosphorothioate-dependent effect similar to previously published data using the TLR7/8 small molecule ligand R-848 (Jurk et al., 2006b; Haas et al., 2008). Suppression of TLR7-mediated GU-rich ORN activation may occur either at the receptor side or in the signaling pathway. In case of small molecules like chloroquine and nucleic acids like CpG ODN, direct binding to TLR9 has been demonstrated (Rutz et al., 2004). Therefore, the postulated theory of one ligand being the activator and the other the regulator (Jurk et al., 2006a) at the TLR side seems to be also feasible for other nucleic acid ligands such as GU-rich RNA. On the other hand, it is possible that a combined TLR7 and TLR9 activation by nucleic acid TLR agonists may be tightly regulated to avoid potential pathological consequences of overstimulation as suggested by others (Liew et al., 2005; Lang and Mansell, 2007). In contrast, the combined stimulation of TLR8 and TLR9 is tolerated due to the activation of divergent cytokines and chemokines. However, 8-oxo-2′-deoxy-guanosine and 8-bromo-2′-deoxy-guanosine modification of the first 5′ guanosine results in an increase in type I IFN production and appears to be due to additive effects of TLR7 and TLR9. Nevertheless, in these experiments not 2 nucleic acid agonists were combined, but a 5′ modification was introduced that was previously described to be a TLR7 agonist. Therefore, 2 TLR agonists different in nature may stimulate the TLRs at different binding sites, and not compete for TLR activation. This may overcome the protection of the immune system against overstimulation.
Our results may be an important contribution to the research on activation and regulation of TLRs, as well as to pharmaceutical research and drug development. We have identified immune stimulatory ORNs that do not require a delivery system for TLR7 or TLR8 activation, as well as ORNs that combine 2 or 3 TLRs: TLR7/TLR8, TLR7/TLR9, or TLR7/TLR8/TLR9. Divergent immune modulatory responses via TLR7, TLR8, or TLR9 and the combination thereof maybe beneficial for different clinical indications such as in cancer, allergy, or infectious diseases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.