
Abstract
Nonstructural protein 1 (NS1) of influenza A viruses counteracts the host immune response against the influenza viruses by not only inhibiting the nuclear export and maturation of host cell messenger RNA (mRNA), but by also blocking the double-stranded RNA–activated protein kinase–mediated inhibition of viral RNA translation. Reduction of NS1 gene product in the host cell may be a potent antiviral strategy to provide protection against the influenza virus infection. We used small interfering RNAs (siRNAs) synthesized against the viral mRNA to down regulate the NS1 gene and observed its effect on inhibition of virus replication. When NS1 gene–specific siRNA were transfected in Madin Darby canine kidney (MDCK) cells followed by influenza A virus infection, approximately 60% inhibition in intracellular levels of NS1 RNA was observed. When siRNA was administered in BALB/c mice, 92% reduction in the levels of NS1 gene expression in mice lungs was observed. A significant reduction in the lung virus titers and cytokine levels was also detected in the presence of siRNAs as compared with the untreated control. The study was validated by the use of selectively disabled mutants of each set of siRNA. Our findings suggest that siRNA targeted against NS1 gene of influenza A virus can provide considerable protection to the virus-infected host cells and may be used as potential candidates for nucleic acid–based antiviral therapy for prevention of influenza A virus infection.
Introduction
Various RNA interference (RNAi) strategies are available for sequence-specific intervention of the target gene expression (Kumar et al., 2010). These include siRNA, ribozymes, DNA-enzymes, aptamers, and antisense DNA or RNA molecules. siRNAs are short (21–26 nucleotides) double-stranded RNAs (Elbashir et al., 2001) that facilitate post-transcriptional gene silencing through a series of steps after the formation of RNA-induced silencing complex (Hammond et al., 2000; NISHIKURA, 2001). siRNAs are naturally produced in the mammalian cells by an enzyme named DICER, a double-stranded (Ds-RNA)–specific endonuclease, which cleaves the long Ds-RNA molecules into short Ds-RNA fragments.
The application of siRNA-mediated RNAi as an antiviral approach was first demonstrated against respiratory syncytial virus (Bitko & Barik, 2001). The authors showed specific degradation of protein and RNA levels of the target genes. Since then, siRNA has been used to down regulate the expression of various genes in different viruses. The first report of siRNAs being used to down regulate the expression of influenza A virus genes was given by Ge and colleagues in 2003 (Ge et al., 2003). An array of siRNAs was designed against NP, PA, PB1, PB2, M, and NS genes of influenza A virus and tested in mammalian cells and 10-day-embryonated chicken eggs. The siRNAs targeted against PB1, PA, and NP genes showed inhibition of influenza A virus at higher efficiency than the siRNAs against PB2, M, or NS. Subsequently, the same siPA and siNP were tested in vivo to study inhibition of different subtypes (H1N1, H5N1, H7N7, or H9N2) of influenza A virus (Ge et al., 2004; Tompkins et al., 2004).
Another strategy of influenza virus replication inhibition includes down regulation of certain cellular genes essential for the virus replication. Various research groups have identified many such genes which can be targeted using siRNA-mediated RNAi technology to facilitate virus inhibition in infected cells (Elton et al., 2001; Satterly et al., 2007). However, since the cellular genes are crucial for the viability of the cells, only some of the genes, which can be partially dispensable for cellular functioning, can be targeted for viral treatment (Aszódi et al., 1999; Harborth et al., 2001; Hoffman et al., 2003).
There have been reports mentioning the suppression of RNAi by certain viral RNAs and proteins. Some of the viral encoded RNAi suppressor proteins include Primate foamy virus (PFV-1) Tas, influenza A virus NS1, hepatitis C virus (HCV) Core, human immunodeficiency virus (HIV)-1 Tat, Vaccinia virus E3L, and the Ebola virus VP35 protein. Viral RNAs like adenovirus virus–associated RNAs 1 and 2 (VA1 and VA2) are also reported as RNAi suppressors (Li et al., 2004; Lu et al., 2004; Andersson et al., 2005; Bennasser et al., 2005; Lecellier et al., 2005; Wang et al., 2006; Haasnoot et al., 2007). The NS1 protein of influenza A virus has been reported as the first human viral protein to inhibit RNAi in plants and Drosophila (Bucher et al., 2004; Delgadillo et al., 2004; Li et al., 2004). However, the NS1 protein–mediated suppression of RNAi is strictly dependent on the type of cell line (Haasnoot et al., 2007). NS1 suppresses the RNAi in HEK 293 T cells (de Vries et al., 2009) but not in HeLa cells (Hang Kok & Jin, 2006).
The effect of siRNA against the nonstructural gene of influenza A virus has been studied before (Ge et al., 2003), but no inhibition was reported by the researchers. In our study, we have shown that siRNA targeted against the NS1 gene of influenza A virus could inhibit NS1 gene expression levels up to 92% in mice. We also showed that the inhibition of NS1 gene of influenza A virus by siRNAs is able to reduce the virus lung titers and provide effective protection to mice from lethal virus challenge.
Materials and Methods
Cells, mice, and viruses
MDCK cells (purchased from NCCS Pune, India) were maintained in minimum essential medium (Sigma, St. Louis, MO) supplemented with 0.3% sodium bicarbonate, 10% fetal calf serum, 100 U/mL of penicillin and 100 μg/mL of streptomycin. Six- to eight-week-old, male BALB/c mice were obtained from the Institute of Nuclear Medicine and Allied Sciences, New Delhi, India, and were housed under specific pathogen-free conditions, as validated by screening of sentinels. All the animal experiments in this study were done as per the guidelines of Indian National Science Academy, India. Experimental use of mice was approved by the Institutional Animal Ethical Committee of Vallabhbhai Patel Chest Institute, University of Delhi.
The influenza virus strain [A/PR/8/34 (H1N1)] used for this study was obtained from Centers for Disease Control, Atlanta, Georgia, and propagated in MDCK cell line. The culture supernatants containing the virus were collected and stored at −70°C for further use.
Designing, synthesis, and annealing of siRNAs
Four sets of siRNAs were designed by using the siDIRECT software against the conserved regions of the NS1 gene (Naito et al., 2004) and were commercially synthesized. Equimolar amounts of sense and anti-sense strands of siRNAs were annealed by incubation at 95°C for 5 minutes and then gradual reduction of the temperature at a rate of 1°C per 30 seconds until a temperature of 35°C was reached, then by 1°C per minute until the temperature reached 5°C. The siRNA duplex formation was confirmed by gel electrophoresis. Sequences of the NS1-specific siRNAs used are as follows:
NS-8: sense 5′-GCAGGGUGACAAAGACAUAAU-3′ antisense 5′-UAUGUCUUUGUCACCCUGCUU-3′ NS-221: sense 5′-GGAGCGGAUUCUGAAAGAAGA-3′ antisense 5′-UUCUUUCAGAAUCCGCUCCAC-3′ NS-421: sense 5′-CGAACUUCAGUGUGAUUUUUG-3′ antisense 5′-AAAAUCACACUGAAGUUCGCU-3′ NS-535: sense 5′-CUGCUGAGGAUGUCAAAAAUG-3′ antisense 5′-UUUUUGACAUCCUCAGCAGUA-3′
Cloning of NS1 gene of influenza A virus
Viral RNA was isolated using QIAamp viral RNA isolation mini kit (Qiagen, GmBH, Hilden, Germany) as per manufacturer's instructions. The NS1 coding sequence was amplified from total viral RNA by using Qiagen's one step reverse transcriptase-polymerase chain reaction (RT-PCR) kit, using specific primers (forward primer: 5′ TTATggtaccGGGAGCAAAAGCAGGGTG 3′ and reverse primer: 5′ ATATctcgagTATTAGTAGAAAACAAGGGTGTTTT 3′). The nucleotides in lowercase indicate restriction sites for Kpn1 and Xho1 in the forward and reverse primers respectively. The NS1 gene was cloned in pSecTag2B vector.
RT-PCR and real-time RT-PCR analysis
MDCK cells were seeded in a 6-well plate at density of 0.5×106 cells/well. The cells, at 70% confluency, were co-transfected with 1 μg of the sequence-confirmed NS1-pSecTag2B plasmid along with various concentrations of siRNA duplexes ranging from 10 pmol to 60 pmol using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) as per the manufacturer's instructions. Mock-transfected and only NS1-pSecTag2B–transfected MDCK cells were used as controls for the experiment. After 48 hours of transfection, total cellular RNA was isolated using Ribozol RNA extraction reagent (Amresco, Solon, OH). The isolated RNA was treated with an appropriate concentration of DNase to avoid any DNA contamination. The concentration of the RNA was determined spectrophotometrically and then 1 μg of RNA from each well was subjected to one-step RT-PCR amplification of NS1 gene for assessment of its expression levels. Simultaneously, murine glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as an internal gene control for each sample. The down regulation in NS1 gene expression was also analyzed by performing real-time RT-PCR using SYBR Green dye (Bio-Rad Laboratories, Hercules, CA) as per the protocol standardized in our laboratory (Kumar et al., 2010). All of the experiments were performed in triplicate.
We assessed the efficacy and specificity of siNS1 by analyzing the effect of irrelevant siRNA (siM1) or mt-siNS1 on NS1 gene expression and effect of wt-siNS1 on the gene expression of M1 gene respectively. MDCK cells treated with 50 pmol wt-siNS1 or mt- siNS1 or siM1 were transfected with NS1-pSecTag2B clone. For specificity analysis, M1-pcDNA3.1 (+) clone with or without 50pm wt-siNS1 was transfected in MDCK cells. Total cellular RNA was isolated, after 48 hours of transfection, as previously described and subjected to SYBR Green I–based real-time RT-PCR analysis (Kumar et al., 2012). The experiment was performed in triplicates and the data was expressed as means±standard deviation (SD).
In vivo experiments
Six- to eight-week-old, male BALB/c mice were used as in vivo models to study the effect of siRNAs on influenza virus replication. Seven animals in each study group were studied as follows.
Group 1: mock-infected and mock-treated group (normal control)
Animals in this group were mock treated and inoculated with 1× phosphate-buffered saline (PBS) via intra nasal route.
Group 2: mock-treated and virus-infected group (virus control)
Animals in this group were given intra nasal instillation with 2×106 plaque-forming units (p.f.u.) of A/PR/8/34 influenza virus diluted in 100μL of sterile 1× PBS. No siRNA was administered to the animals in this group.
Group 3: siRNA-treated and virus-infected group
This group was further subdivided into 4 subgroups. Animals under the subgroups 1, 2, 3, and 4 were administered 10 μM, 20 μM, and 30 μM of wild-type (wt) siRNA and 30 μM of mutant (mt) siRNA respectively via intravenous route (Ibrahim et al., 2011). Tweny-four hours post injection, mice were given another dose of siRNAs by intranasal route to enhance the efficacy of siRNAs in lungs. After 4 hours of siRNA delivery, mice were instilled with 2×106 p.f.u. of A/PR/8/34 influenza A virus. Mice were sacrificed on day 2 post virus infection for collection of bronchoalveolar lavage fluid (BALF) and lungs. The delivery of siRNAs in mice was done using in vivo jet polyethylenimine (PEI) reagent (HiMedia Laboratories Pvt. Ltd.) as per the manufacturer's instructions.
Real-time RT-PCR analysis
The mice lungs were collected in RNAlater (Sigma) and homogenized for isolation of total RNA using RNeasy kit (Qiagen) as per the manufacturer's protocol. The lung RNA samples were quantified spectrophotometrically. One μg of each RNA sample was subjected to RT-PCR for assessment of NS1 RNA levels. Gene coding for mouse GAPDH was also PCR amplified to serve as an internal gene control.
For further quantification of NS1 RNA levels in all the groups, SYBR Green–based real-time RT-PCR analysis was performed using an iScript one-step RT-PCR kit (Bio-Rad Laboratories) as described previously (Kumar et al., 2010).
SDS-PAGE and immunoblotting
The lungs of mice were immediately frozen by incubation in liquid nitrogen. The frozen lung samples were homogenized into powder and incubated in chilled mammalian cell lysis buffer [containing 0.1M NaCl, 0.01M Tris-Cl (pH 7.6), 0.001M EDTA (pH 8.0), 1 mM of protease inhibitor cocktail (Amresco), and 1mM of phenylmethylsulfonyl fluoride] for 30 minutes. The suspension was collected in sterile tubes and centrifuged at 13,000 rpm for 30 minutes at 4°C. The supernatant containing the protein was aliquoted in fresh tubes and stored at −20°C for analysis by SDS-PAGE and immunoblotting. After determination of protein concentration by Bradford method (ProPure Protein assay kit, Amresco), the samples (40 ng/lane) were fractionated on 12% polyacrylamide gel followed by western blotting using primary (1:100 dilution) goat monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) against NS1 protein of influenza A virus and horseradish peroxide conjugated bovine–anti goat immunoglobin G (1:1000 dilution) as secondary antibody. The extracts prepared from lungs of only virus-infected mice were taken as positive control and untreated/uninfected mice were taken as negative control.
Virus quantitation in mice lungs
The lungs of mice from all the groups were homogenized in PBS containing 1% bovine serum albumin (BSA) and centrifuged at 3,000 rpm for 15 minutes at 4°C. Ten-fold dilutions of lung homogenates were prepared in PBS-containing 1% BSA and used for plaque assay as described earlier (Huprikar et al., 1980). The results were expressed as mean p.f.u. per mL+SD.
Histopathological examination of lung tissue
At day 2 post infection, lungs were collected and fixed with 10% neutral buffered formalin solution. Following fixation, lungs were embedded in paraffin and 5 μm sections were cut. Sections were stained with hematoxylin and eosin and examined for inflammatory features.
Assessment of cytokine levels in bronchoalveolar lavage fluid
The lungs of mice were perforated using 100 μL of PBS through the tracheal bifurcation for collection of bronchoalveolar lavage fluid. BALF from all the mice groups was aliquoted and stored at −80°C until further use for assessment of cytokine levels. Interferon (IFN)-γ, tumor necrosis factor (TNF)-α, IFN- α1, IFN-β, and interleukin (IL)-1β were determined using cytokine ELISA kits (BD Pharmingen, BD Biosciences, San Jose, CA) as per manufacturer's protocol.
Survival assay
The BALB/c mice (n=6 for each study group) were administered siRNA and instilled with influenza A virus as described earlier in the section “In vivo studies.” The mice were monitored twice daily up to 21 days post infection for their survival and to analyze the protective effect of siRNA against the influenza A virus infection.
Statistical analysis
The data were analyzed by Student's t test and one-way analysis of variance and all the results were expressed as mean+SD.
Results
Down regulation of NS1 gene expression in MDCK cells
The NS1-pSecTag2B clone was commercially sequenced for confirmation of the NS1 coding sequence in correct orientation. NS1 clone and siRNA co-transfected MDCK cells were harvested 48 hours post transfection for analysis of intracellular levels of NS1 gene. Out of the 4 sets of siRNAs, 2 were found to be effective against the target gene. A decrease in RNA levels of NS1 gene was observed with increasing concentration of transfected siRNA. The siRNAs that worked best were NS-221 and NS-421. The results observed with both the siRNA sets were almost comparable. The data shown in this paper represent the results observed using siRNA NS-221. Approximately 60% inhibition in NS1 gene expression was observed using 50 pmol of siRNA as observed by RT-PCR (Fig. 1a) and real-time RT-PCR analyses (Fig. 1b). The result was found to be constant at concentration of 60 pmol and higher of wt-siNS1. The experiments were performed in triplicate and the error bars in the figures indicate standard deviation. The inability of the irrelevant siRNA (i.e., siM1) against M1 gene or mutated siNS1 to down regulate the expression of NS1 gene confirmed the efficacy of wt-siNS1 (Fig. 1c). In order to further validate the specificity of siNS1 designed by us, we analyzed the effect of wt-siNS1 on the gene expression of M1 gene, used as unrelated viral RNA control. The level of M1 RNA in the 50pm wt-siNS1–treated and M1-pcDNA3.1 (+) –transfected MDCK cells was almost similar to the RNA levels in the MDCK cells transfected with the M1-pcDNA3.1 (+) clone alone (Fig. 1d). This showed that the siRNAs were specific for the NS1 gene and efficiently inhibited the target gene expression.
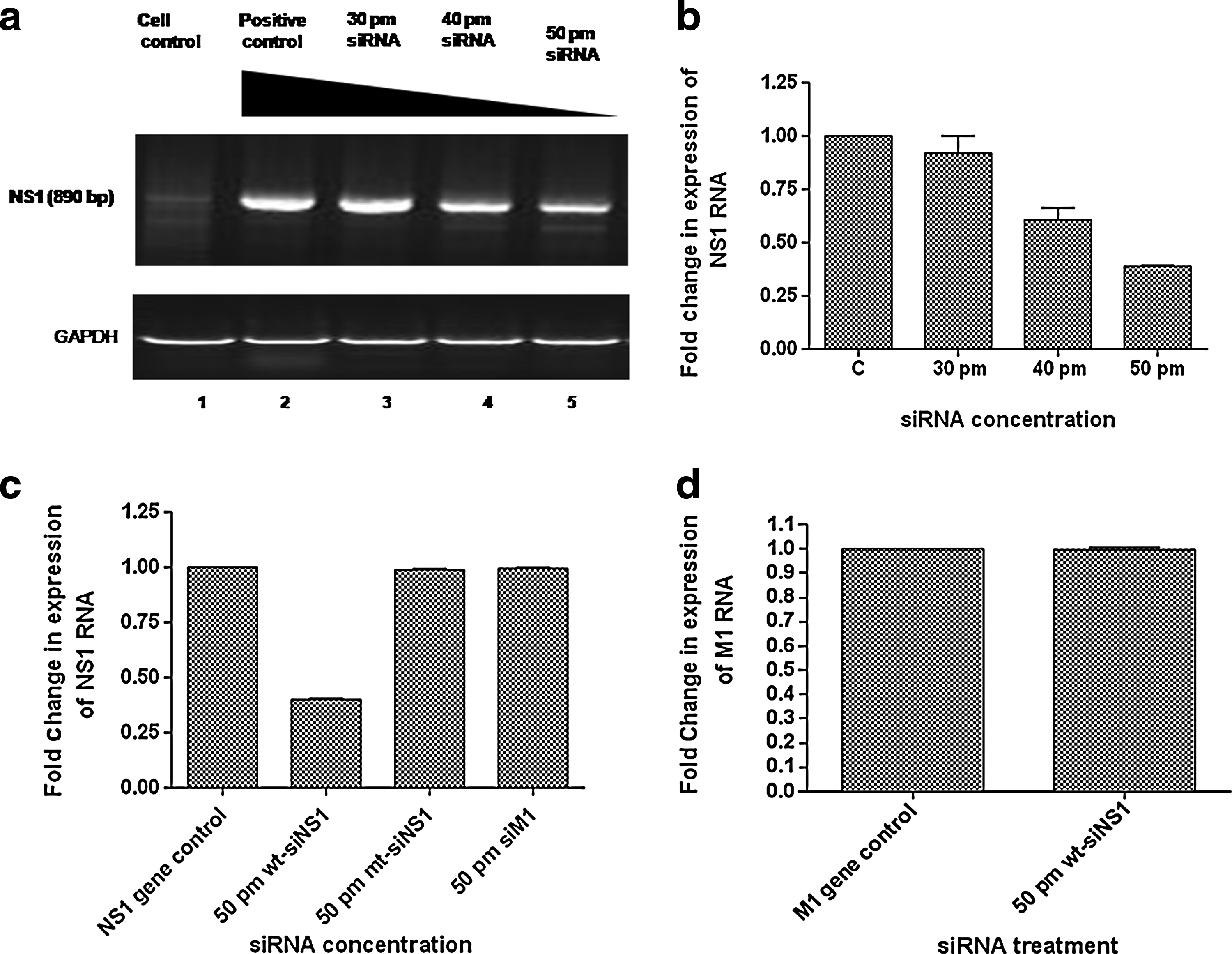
RT-PCR analysis shows intracellular reduction of nonstructural protein 1 (NS1) RNA in Madin Darby canine kidney (MDCK) cells treated with wild-type small interfering RNA (wt-siRNA).
Lung virus titers in BALB/c mice
The potent effect of siRNAs was also validated by assessment of virus titers in lungs of siRNA-treated mice. The mice were given intravenous injections of siNS1 using in vivo jet PEI reagent. The virus titers were significantly reduced in the lungs of mice treated with 30μM of wt-siRNA (Fig. 2) as compared with mt-siRNA and control mice groups. The use of mt- siRNA allowed us to validate the siRNA-specific inhibition in lung virus titer. The experiment was performed in triplicate and the error bars in Fig. 2 indicate SD.
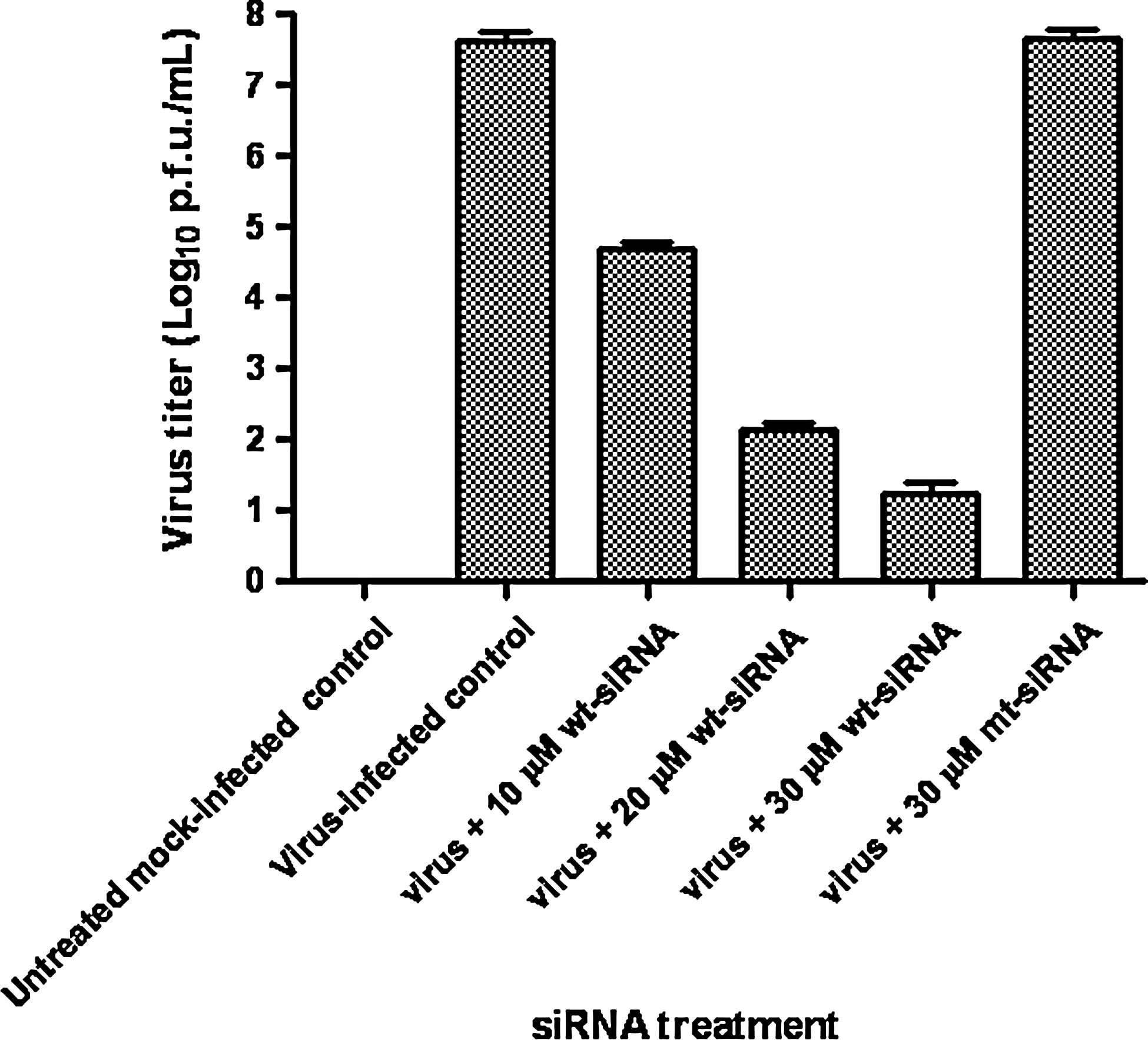
The mice lungs were collected 2 days post virus challenge and homogenized. Virus titration was performed on lung homogenates by plaque assay. The virus titer in the lungs of mice treated with different concentrations of wt-siRNA was significantly reduced in comparison with virus-infected control or mutant siRNA groups. The experiment was performed in triplicate and the error bars indicate SD.
Decreased NS1 expression levels in siRNA-treated mice lung
The level of NS1 RNA from lung homogenates in different groups was analyzed by SYBR Green–based real-time RT-PCR. The ΔΔCt values were calculated and compared with the RNA level of NS1 in lungs of untreated virus–infected mice. It was observed that the expression of NS1 RNA decreased up to 92% in mice treated with 30μM siRNA (Fig. 3a). The experiments were performed in triplicate and the error bars indicate SD.
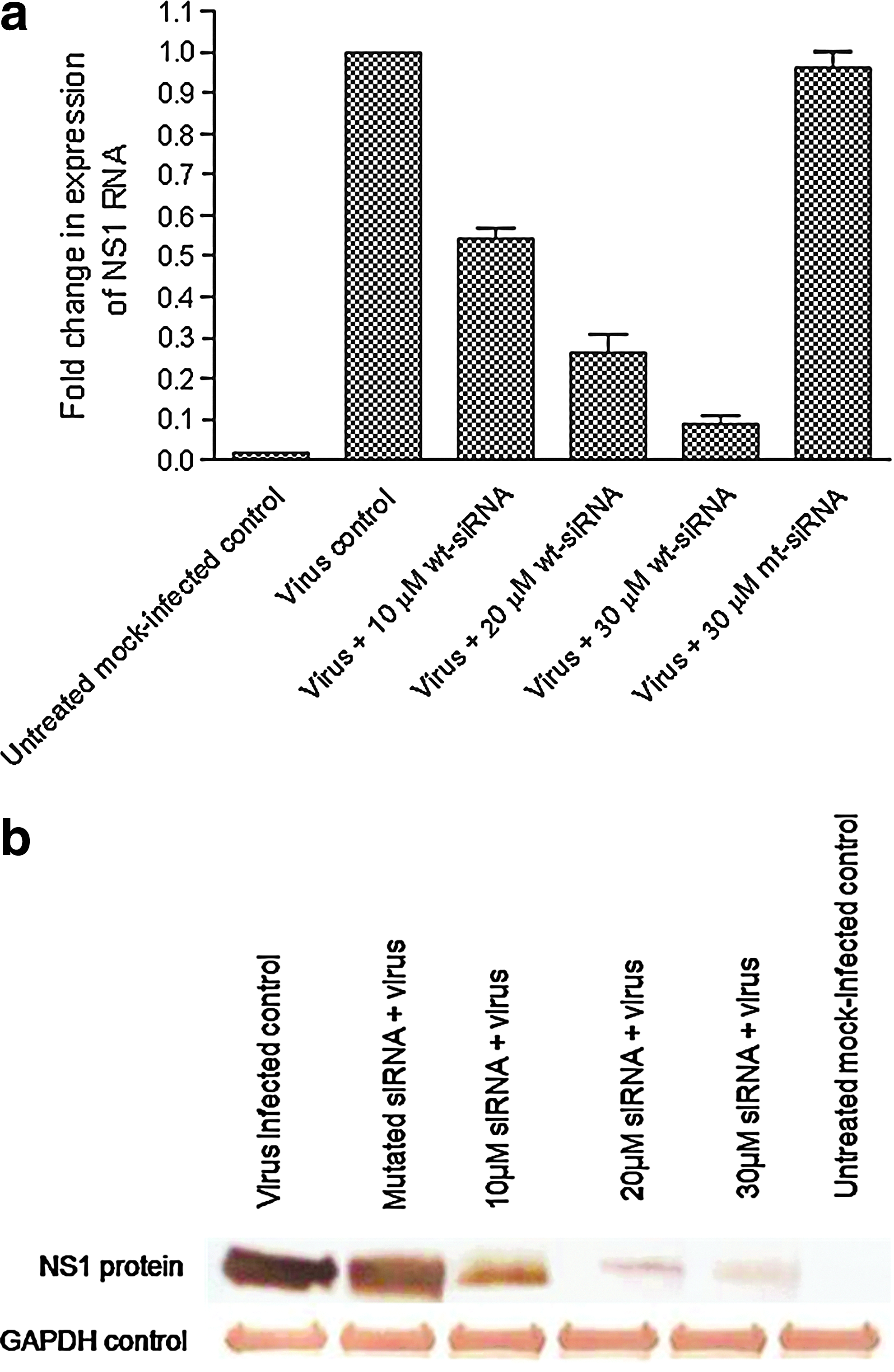
Different concentrations of wt-NS1–specific siRNA were administered via intravenous injection in mice followed by additional dose of wt-siRNA 4 hours before virus challenge. On day 2 post virus challenge, mice lungs were harvested for RNA isolation and cell lysate preparation.
Down regulation of NS1 gene at protein level was checked in terms of inhibition of protein expression as observed by western blotting. The NS1 protein expression was reduced by 90% in 30-μM siRNA-treated groups as compared with virus-infected and mutant groups (Fig. 3b). The observations at both RNA and protein levels suggest effective inhibition of NS1 gene expression in a dose-dependent manner. The data shown is from 1 of 2 experiments.
Cytokine analysis
The BAL fluid of the mice from each group was analyzed for virus-specific cytokines, viz. TNF-α, IFN-γ, IFN-α1, IFN-β, and IL-1β. We also verified the absence of any siRNA induced nonspecific antiviral responses by testing the BALF samples of only siRNA-treated mice for cytokine profiling. The pattern of cytokine expression in the lungs of infected mice showed the modulation of virus replication in the presence of different concentrations of siRNA (Fig. 4). The levels of TNF-α and IFN-γ reduced by 15- to 20-fold upon treatment with 30 μM wt-siNS1 in comparison with the untreated virus control, whereas the levels of IFN-α1, IFN-β, and IL-1β were relatively higher in wt-siRNA–treated mice in comparison with the virus control or mt-siRNA–treated mice group. There was no significant difference observed in cytokine levels of virus-infected group and the mt-siRNA group. This showed that the inhibition in the level of cytokine production was due to the effect of the siRNA against the virus infection in mice. Values are means±SD (n=7 per group).
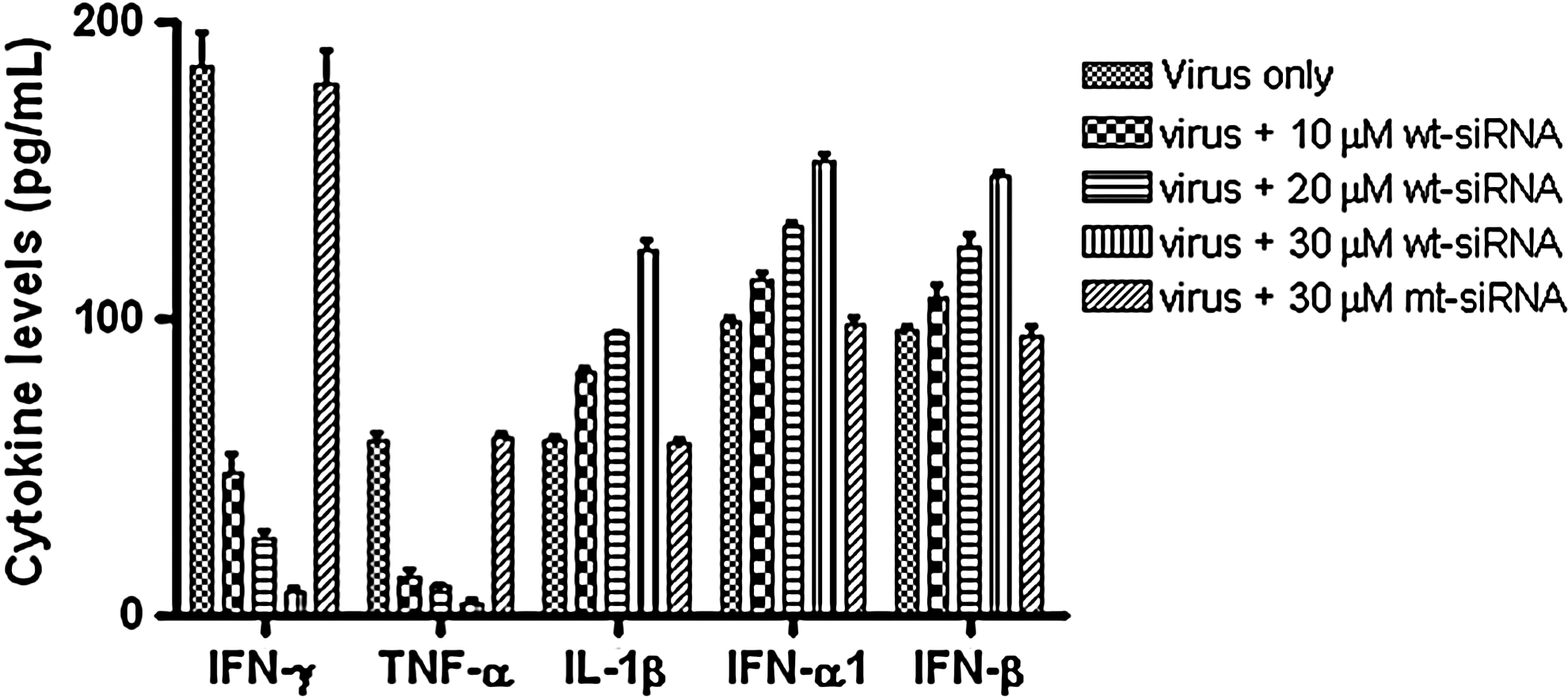
The bronchoalveolar lavage (BAL) fluid was collected 2 days post virus challenge and assessed for IFN-γ, TNF-α, IFN-α1, IFN-β, and IL-1β by ELISA. A considerable inhibition in the Interferon (IFN)-γ and tumor necrosis factor (TNF)-α, levels of wt-siRNA–treated mice was observed, while there was no significant difference in the cytokine levels of virus control group mice and mt-siRNA–treated mice. The values of IFN-α1, IFN-β, and interleukin (IL)-1β were relatively higher in wt-siRNA treated mice in comparison with the virus control or mt-siRNA treated mice group. Values are means±SD (n=7 per group).
Histopathological studies
Sections from virus-infected control lungs revealed damage by influenza virus with bronchial inflammation and peribronchial lymphoid aggregates. The alveoli were filled with cell debris and fluid, pneumocyte hyperplasia was seen in the alveolar walls. A significant reduction in inflammatory damage was noted with increasing dose of wt-siRNA (NS-221). However, no significant reduction was observed on treatment with mt-siRNA (Fig. 5). The use of mt-siRNA validated the efficacy of NS1-specific siRNA. The data shown is representative of the 2 experiments.
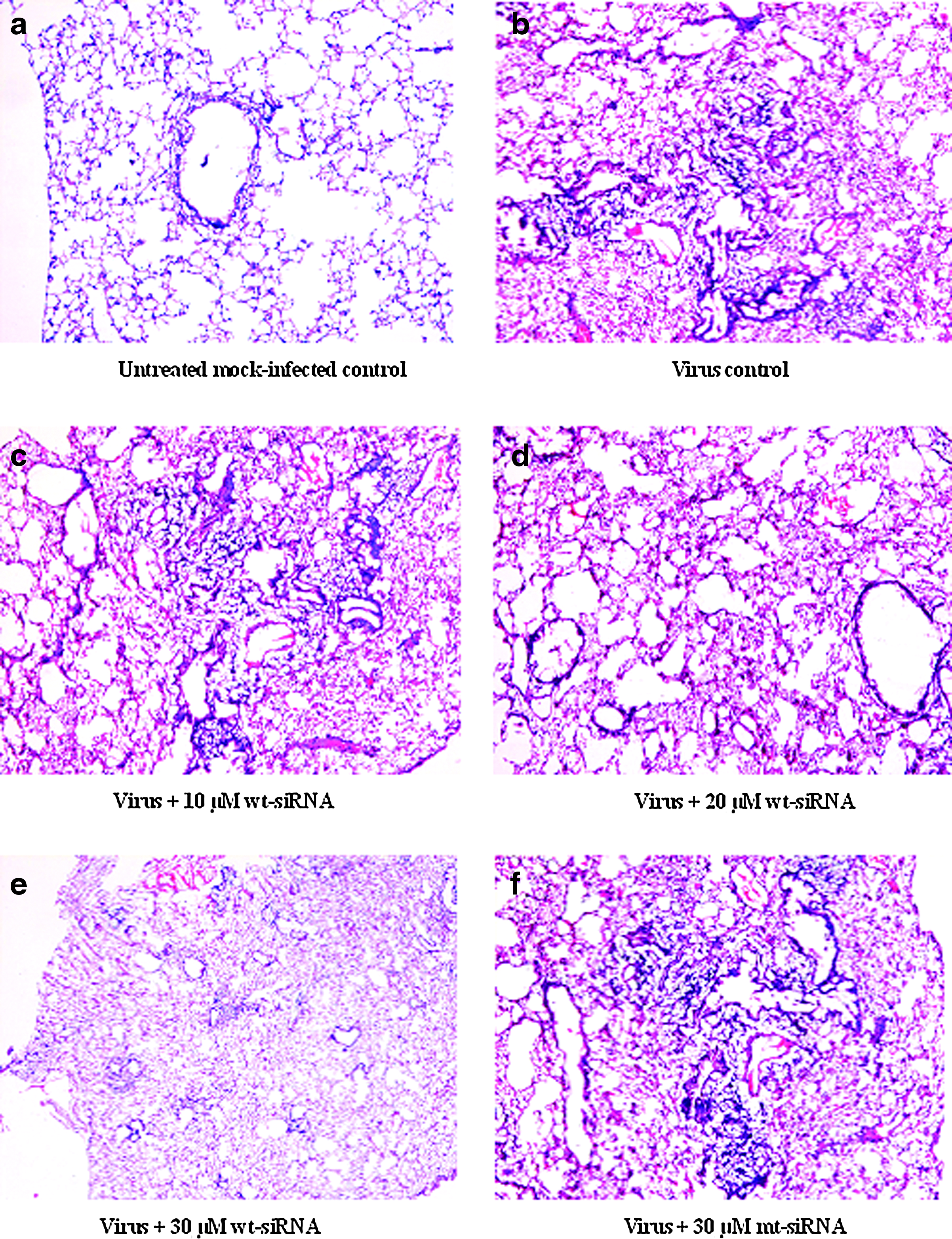
Photomicrograph showing haematoxylin & eosin stained mouse lung tissue samples (100× magnification).
Animal survival assay
Mice survival assay was also performed to confirm whether the siRNA is able to protect the animals from lethal influenza virus dose. On day 16 post influenza virus challenge, only 40% animals survived in the virus control or mt-siRNA group, whereas in the wt-siRNA (30 μM) group, 100% animal survival was observed during the 21-day period of the assay (Fig. 6). The protection provided by NS1-specific siRNA treatment was statistically significant (P<0.001).
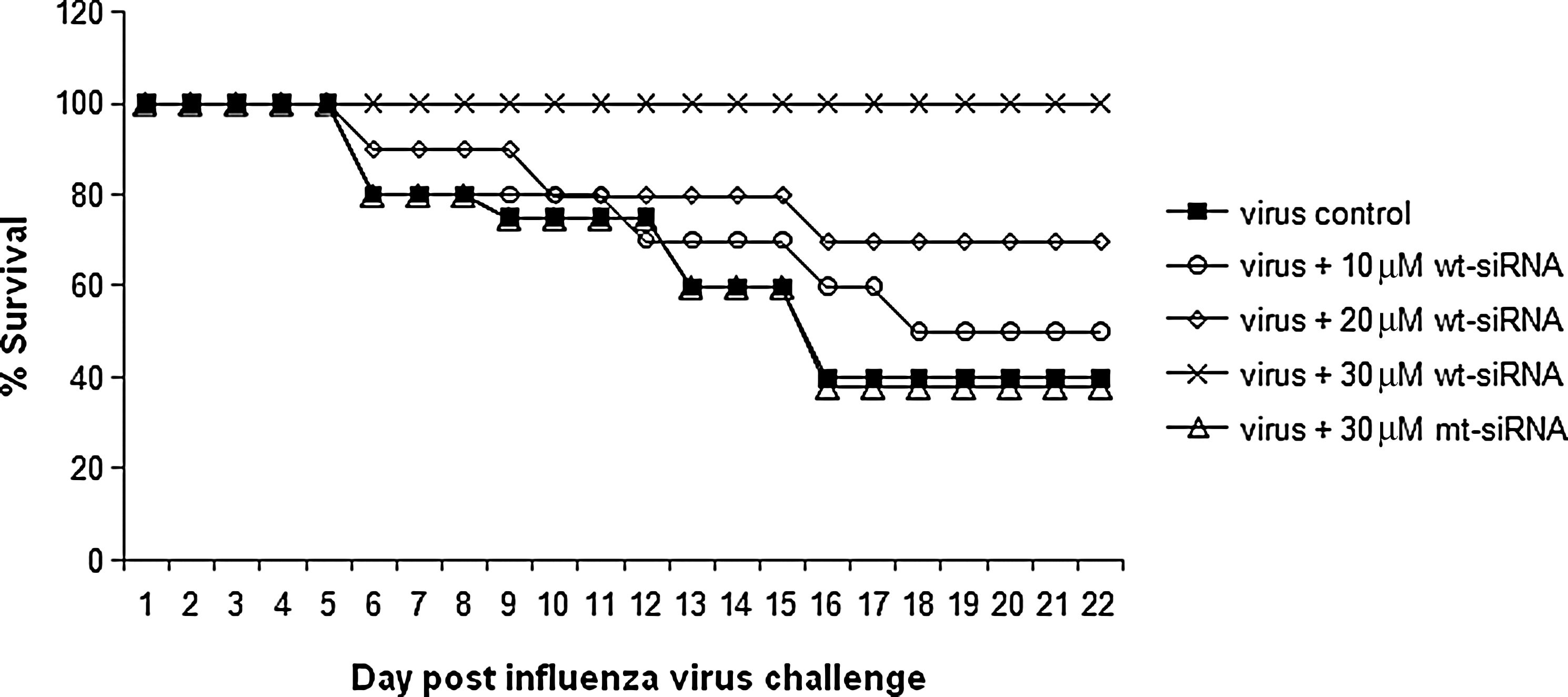
Male BALB/c mice (n=6 for each study group) were treated with various concentrations (10 μM, 20 μM, and 30 μM) of wt-siRNA and 30μM of mt-siRNA followed by lethal A/PR/8/34 virus challenge. The mice were monitored twice daily for 21 days. There was significant reduction in the mice mortality rate in wt-siRNA–treated virus-infected mice groups. The protection provided by NS1-specific siRNA treatment was statistically significant (P<0.001).
Discussion
The nonstructural gene of influenza A virus is known for its protective functions which work in the favor of the viruses for their replication and propagation in host cells. The need for an annual influenza vaccine formulation, owing to the continuous mutation in its genome, calls for an effective strategy that can restrain the virus replication in host cells. We have previously shown potent inhibition of influenza A virus replication in a mammalian cell line using siRNA-chimeric-Rz constructs (Kumar et al., 2010) or DNAzymes (Kumar et al., 2011) targeted against the conserved matrix gene. In our present study, we showed that the siRNAs targeted against the nonstructural gene of influenza A virus could effectively suppress virus replication. We designed synthetic siRNAs that could be successfully delivered to the host cells both ex vivo and in vivo to bring specific inhibition of the influenza A virus NS1 gene. Different modes of in vivo delivery of siRNAs previously employed by researchers include oligofectamine (Tompkins et al., 2004) or PEI reagent (Ge et al., 2004). For our study, we administered siRNAs via intravenous route using in vivo jetPEI reagent, which showed efficient delivery of the siRNA in mice.
Our results are in accordance with the RNAi studies using siRNAs as potent antiviral agents (Ge et al., 2003; Ge et al., 2004; Hui et al., 2004; Tompkins et al., 2004) for treatment of viral infection in animal model. We analyzed the efficacy of siRNAs against the NS1 gene of influenza A/PR/8/34 (H1N1) virus. The study was conducted with both wild-type and mutant siRNAs to elucidate that the NS1 gene suppression was highly specific and that the viral gene expression could be modulated accordingly. Our study revealed that the designed siRNAs could not only suppress the NS1 transcript but also conferred significant protection against the lethal virus challenge by the A/PR/8/34 (H1N1) strain.
The NS1 protein has also been reported to inhibit the synthesis of type 1 IFN (Mibayashi et al., 2007) and inflammatory cytokines by blocking retinoic acid inducible gene-1 (RIG-1) activation (Guo et al., 2007; Opitz et al., 2007; Pachler and Vlasak, 2011). RIG-1 is a key regulator of antiviral immunity which is activated by the single-stranded viral RNA containing phosphate group at 5’-end. The interaction of RIG-1 with the viral RNA initiates a signaling cascade, leading to the production of IFN. But, ubiquitination of RIG-1 near its amino terminus (known as the CARD domain) is necessary for induction of pathways leading to IFN production. It has been shown that NS1 protein of influenza virus inhibits the ubiqutination of RIG-1 by tripartite motif –containing protein 25 (TRIM25), by binding to TRIM25 and preventing it from forming multimers (Gack et al., 2009). A recent study shows that the RIG-1-dependent IFN-β induction by the 5′ UTR sequences of the influenza cRNA is dependent on the presence of a 5′-phosphate group (Davis et al., 2012). We observed that the levels of TNF-α, IFN-γ in mice decreased by 15- to 20-fold upon treatment with 30μM wt-siNS1 as compared with untreated virus controls. This indicates that the wt-siNS1 used in our study effectively suppressed virus replication in mice lungs, thereby leading to a decrease in the cytokine response. Our observation, that the IFN-α1, IFN-β, and IL-1β levels were higher in wt-siRNA–treated mice as compared with the virus control or mt-siRNA–treated mice group, supported the previous reports (Mibayashi et al., 2007; Jia et al., 2010; Sharma et al., 2011) that the NS1 protein suppresses the production of IFN-α1, IFN-β, and IL-1β.
The histopathological analysis of mice lungs treated with wt-siRNA showed significant reduction in the bronchial inflammation and peribronchial lymphoid aggregate formation, thereby providing evidence for the potent effect of wt-siRNA against influenza A virus replication (Fukushi et al., 2011).
In light of earlier reports regarding the RNAi suppression activity of NS1 protein of influenza A virus, we checked the effect of NS1 protein of influenza A/PR/8/34 (H1N1) virus on siRNA-mediated RNAi in our experimental setup. We analyzed the potency of siRNA against M1 gene in the presence of NS1 protein in mammalian cells as well as BALB/c mice. No significant difference in the levels of M1 RNA levels was observed in the presence or absence of NS1 protein both under ex vivo or in vivo conditions. It was consistent with the previous findings that the RSS activity was exhibited by only some of the NS1 proteins (Haasnoot et al., 2007; de Vries et al., 2009) and the RNAi suppression is host-cell dependent.
We concluded that the efficient suppression of the NS1 gene transcript paralleled a highly significant decrease in virus replication. Thus, the siRNAs tested in our study may be used in the prevention of influenza A virus infection, and these molecules may be used as effective antiviral drug candidates.
Footnotes
Acknowledgments
We thank the Council of Scientific and Industrial Research, Government of India, for providing the financial support to the authors for successful completion of this research work. We also thank Amitabh Mathur and Alivia from Department of Zoology, University of Delhi for helping with mice lung histopathology analysis.
Author Disclosure Statement
No competing financial interests exist.