
Abstract
Investigation of the intracellular fate of small interfering RNAs (siRNAs) following their delivery into cells is of great importance to elucidate their dynamics in cytoplasm. Here we describe the use of an advanced fluorescence-based method to probe the dissociation and/or degradation of double-labeled siRNAs in HeLa cells in comparison with that in human embryonic kidney 293T (HEK293T) cells. This work was performed with three siRNAs labeled with fluorescence resonance energy transfer (FRET) dyes, allowing a non-destructive and non-invasive assessment of the dissociation and degradation state of siRNAs in cultured cells. Our FRET analysis not only shows the asymmetric degradation as well as the time-dependent dissociation of each siRNA strand during the measured time period, underlining the high intrinsic nuclease resistance of duplex siRNAs, but also reveals the longer sustainability of siRNAs in HeLa cells compared with that in HEK293T cells, explaining the gene silencing in HeLa cells is more efficient than that in HEK293T cells. In addition, our single-molecule FRET assays demonstrate the potential of the delineated fluorescence-based technique for future research on biological behavior of siRNAs even at the single-molecule level. The fluorescence-based method is a straightforward technique to gain direct information on siRNA integrity inside living cells, which can provide a detection tool for dynamics of biological molecules.
Introduction
By monitoring the expression of a target gene of the siRNA, several groups were able to assess the potency and duration of siRNA effects, including a number of studies that focused on determining whether chemically modified siRNAs are more potent than unmodified siRNAs (Amarzguioui et al., 2003; Braasch et al., 2003; Chiu and Rana, 2003; Czaudema et al., 2003; Layzer et al., 2004; Bartlett and Davis, 2007). Among them, one study, however, has shown that enhanced intracellular nucleolytic stability is not necessarily correlated with increased duration of the silencing effect (Bartlett and Davis, 2007). In fact, the authors found that silencing in non-dividing cells persisted for up to 1 month from a single dose of an unmodified siRNA, suggesting that siRNAs may be quite stable inside the cell. In this regard, many groups have addressed questions of intracellular siRNA stability and localization by introducing fluorophore-modified siRNAs into live cells and using various microscopy techniques (Ohrt and Schwille, 2008), because the ability of an RNA molecule such as siRNA to persist in the cell among a plethora of ribonucleolytic activities is based on the tightly regulated relative rates of its synthesis and decay (Zamore et al., 2000; Elbashir et al., 2001a; Liu and Paroo, 2010). According to the literature, siRNAs are actively exported from the nucleus (Ohrt et al., 2006), except in cases where the RNA target is located in the nucleus (Berezhna et al., 2006). Fluorescence fluctuation spectroscopy has been utilized in a separate study to assess the integrity of labeled intracellular RNAs, revealing that doubly labeled RNA suitable for fluorescence resonance energy transfer (FRET) measurement between the fluorophores is relatively unstable in single-stranded form compared to the corresponding siRNA duplex (Raemdonck et al., 2006). Intracellular FRET imaging of double-stranded RNAs has also been employed to show that intact siRNA duplexes accumulate in cellular foci identified as P-bodies (Järve et al., 2007; Jagannath and Wood, 2009). FRET labeled single-stranded RNAs have been used to show that secondary structure in general attenuates degradation in human cell extracts (Uhler et al., 2003). The efficiency of transcript knockdown by siRNA, however, remains unsolved, and the inability to derive rate constants with a convenient technique for directly monitoring RNA degradation has limited the introduction of predictive mathematical models, while regulation of specific mRNA turnover has long been intensively studied as described (Zamore et al., 2000; Elbashir et al., 2001a; Liu and Paroo, 2010).
To better understand the potential of siRNAs in gene therapeutics, a real-time characterization of their degradation kinetics under intracellular and extracellular conditions is necessary. Such an assay should include rapid and precise assessment of RNA stability and should eventually be amenable to high-throughput screening for optimizing siRNA drugs. In this important regard, we recently developed and reported a FRET-based method using oligonucleotide probes in order to study intracellular dissociation (or melting) and sustainability of siRNAs in live human embryonic kidney 293T (HEK293T) cells (Shin et al., 2011), where the FRET probes were specifically designed to observe intracellular dissociation (or melting) and degradation of short synthetic RNAs in real time, thus providing the desired kinetic information in the cells. In the present study, we describe on employment of the developed method in a similar fashion to HeLa cells, which are also known as siRNA-transfectable as HEK293T cells, in order to see whether there is any correlation between transcript knockdown efficiency and siRNA decay kinetics. As illustrated in Fig. 1, we have used a set of doubly fluorophore-labeled RNAs [more precisely, 3 kinds of dye-labeled siRNAs against enhanced green fluorescent protein (eGFP) expression: (1) 5′ sense strand Cy5 label and 3′ antisense 5′-tetramethylrhodamine (TAMRA) label, both-labeled siRNA; (2) 5′ sense strand Cy5 label and 3′ sense TAMRA label, sense-labeled siRNA; and (3) 5′ antisense strand Cy5 label and 3′ antisense TAMRA label, antisense-labeled siRNA] in order to probe in real time the stability of siRNAs and related molecules by FRET, because these probes have been designed such that energy can be transferred from TAMRA to Cy5 when TAMRA and Cy5 are close to each other.
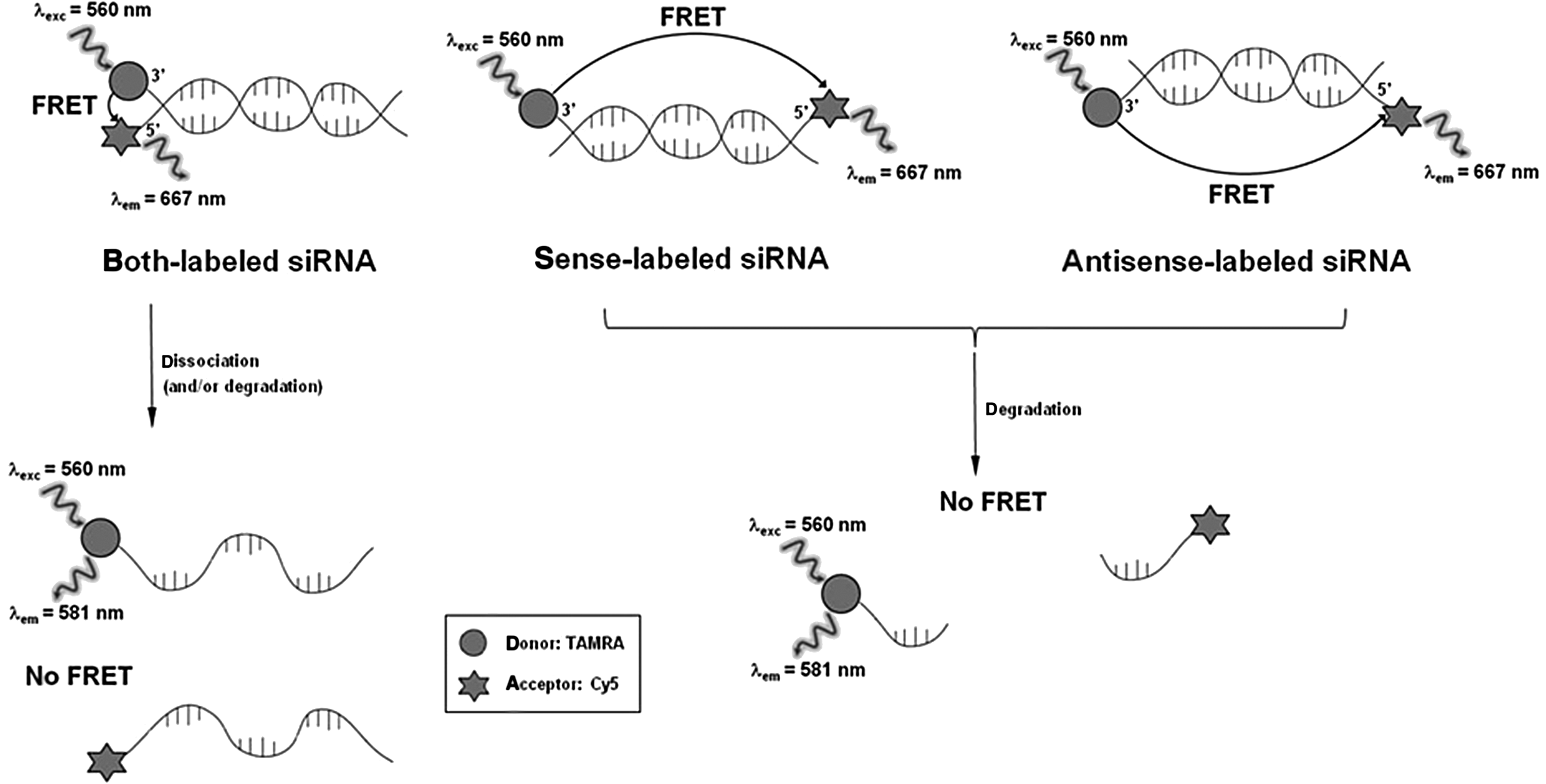
Design and spectral properties of dye-labeled small interfering RNAs (siRNAs), showing fluorescence resonance energy transfer (FRET) and its applicability to probe the dissociation and/or degradation of double-labeled siRNAs. It should be noted that 5′-tetramethylrhodamine (TAMRA) is attached to the 3′-end of the guide stand of both- and antisense-labeled siRNAs and to the 3′-end of the passenger strand of sense-labeled siRNA.
Materials and Methods
Materials
Unless otherwise noted, reagents were obtained from commercial suppliers and were used without further purification. Twenty-one–nucleotide single-stranded siRNAs against eGFP [sense or passenger strand: 5′-pGGC UAC GUC CAG GCG CGC ACC-3′, and antisense or guide strand: 5′-pUGC GCU CCU GGA CGU AGC CUU-3′ (Novina et al., 2002)] were synthesized and purified by Bioneer (Daejun, Korea). Double fluorescent labeling occurred at the 3′ and 5′ ends of the sense and antisense strands with TAMRA and Cy5 respectively; the structures of the double-stranded siRNA derivatives conjugated to the 5′ and/or 3′ of the passenger and guide strands are shown in Fig. 1. The concentrations of the siRNA stock solutions were calculated from absorption measurement at 260 nm (1 OD260=40 μg/mL) with an Agilent 8453 spectrometer, where the absorption of the TAMRA label at 260 nm was taken into account. The presence of the fluorescent labels was verified by absorption measurements at 560 nm for TAMRA [ɛTAMRA(560 nm)=91,000 M–1cm–1] and 650 nm for Cy5 [ɛCy5(650 nm)=250,000 M–1cm–1] (Shin et al., 2011). Three double-labeled siRNA duplexes were prepared through hybridization of antisense and sense RNA strands (Fig. 1). Equimolar amounts of both RNAs were mixed in an annealing buffer (final buffer concentration: 50 mM Tris at pH 7.5, 100 mM NaCl), and annealing was performed in a thermal cycler (2 minutes incubation at 94°C, followed by slow cooling to 25°C over a time period of 45 minutes). Emission fluorescence spectrum of each prepared dye-labeled siRNA was scanned using Synergy Mx fluorimeter upon excitation at 560 nm.
Cell culture
Culture reagents were purchased from Invitrogen. HeLa cells were obtained from American Type Culture Collection and cultured in minimum essential media (MEM) containing 10% fetal bovine serum, 10 U/mL penicillin, and 0.01 mg/mL streptomycin. All cells were cultured at 37°C under a humidified atmosphere of 5% CO2 in air, and exponentially growing cells were cultured following standard procedures until approximately 50% confluence was reached. Cells (approximately 50,000) were plated and harvested by means of trypsinization (1 mL of 0.05% trypsin solution for 1 minute at 37°C), and the resulting single-cell suspension was plated in 6-cm wells at 1×105 cells per well for 12–16 hours prior to eGFP expression vector (pEGFP-N1, BD Clontech) according to the standard transfection protocols. Briefly, cationic lipid complexes were prepared by incubating 0.4 μg eGFP vector with 4 μL lipofectamine (Gibco-Invitrogen,) in 100 μL serum-free MEM (Gibco-Invitrogen) for 20 minutes and were slowly added to the wells. After 4-hour incubation, media were changed with the fresh growth media, and the cells were further incubated. At 16 hours after transfection, eGFP expression was analyzed using fluorescence microscopy in the fluorescein isothiocyanate channel. In other cases, after the16-hour incubation, cells were washed and used for transfection with non-labeled or dye-labeled siRNAs for confocal image acquisition. The expression levels of eGFP and the fluorescence intensities in cells were quantified using Gel-Pro Analyzer (Media Cybernetics).
Confocal images
HeLa cells were seeded in an 8-well LabTek chamber slide (Sigma) at a density of 5×104 and maintained for 24 hours. Liposome–siRNA complexes were prepared in 50 μL serum-free MEM by incubating 30 pmol of double-labeled siRNA with 0.75 μL lipofectamine for 20 minutes at room temperature. After serum-free media were added to the solution, cells were transfected with the complexes and incubated for additional 30 minutes, 1.5 hours, 2.5 hours, and 3.5 hours, respectively. After the transfection, the media were discarded and the cells were washed 3 times with phosphate-buffered saline solution. After the cells were fixed with 3.7% paraformaldehyde solution, confocal images were obtained at each time point using a LSM510 confocal laser scanning microscope (Carl Zeiss Inc.). To acquire an eGFP fluorescence signal (emission: 509 nm), each sample was excited at 488 nm. A fluorescence signal of TAMRA-siRNA was detected at 581 nm with an excitation at 543 nm, and a fluorescence signal of Cy5-siRNA was detected at 667 nm with an excitation at 633 nm, respectively. A FRET signal was obtained at 667 nm with an excitation at 543 nm.
Total internal reflection–based single-molecule FRET measurement
Single-molecule fluorescence detection was performed by a total internal reflection (TIR)-based single-molecule FRET (smFRET) instrument (Roy et al., 2008; Kim et al., 2012a, 2012b). Our system was constructed around a commercial inverted microscope (Zeiss), equipped with a 532-nm diode-pumped solid-state laser system (Nd:YAG continuous crystal laser, 50 mW) and electron-multiplying charged-couple device (CCD) camera (iXon, Andor Technology). The fluorescence signals from TAMRA and Cy5 that were collected by a water-immersion (prism-type) objective lens went through a long-pass filter to block out laser scattering, and the signals lower and higher than 611 nm were separated by a dichroic mirror and detected by a CCD camera with up to 0.2-second time resolution. The observation area was 25 μm×50 μm.
Single-molecule fluorescence detection
Single-molecule fluorescence detection was carried out by adapting literature procedures (Roy et al., 2008; Kim et al., 2012a, 2012b). For a chamber slide preparation, trypsinized HeLa cells were incubated at 37°C with lipofectamine 2,000 and dye-labeled siRNAs (1 pmol) for transfection and deposition on the naked glass surface for 30 minutes. After washing the surface with phosphate-buffered saline, the chamber slide was assembled with a coverslip. For the single-molecule fluorescence detection using the TIR-based smFRET instrument, the chamber slide was mounted on the inverted microscope as reported (Roy et al., 2008; Kim et al., 2012a, 2012b). The resulting fluorescence signals were recorded in real time with a resolution of 0.2 seconds/scan using home-written Visual C++ software (Microsoft). The software was used to obtain each frame of the movie from the camera, and the single-molecule traces were extracted from the recorded movie file using scripts written in IDL (Research Systems).
Results and Discussion
Comparison of gene-silencing activity of differently labeled siRNAs
We first sought to confirm whether the labeling of fluorescent dyes at the 5′ or 3′ end of siRNAs had a minimal effect on gene-silencing activity in HeLa cells and to compare the gene-silencing functionality of dye-labeled siRNAs in HeLa cells. To do this, we performed an eGFP reporter gene-silencing assay for cells transfected with 30 pmol of each of 3 dye-labeled siRNAs (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat). Each siRNA was transfected into the eGFP-expressing HeLa cells that had been transfected with eGFP expression vector and grown for 24 hours. The eGFP fluorescence resulting from the inhibition of eGFP expression was compared with that from the cells transfected with the vector only and the non-labeled standard siRNA to confirm the efficacy of gene silencing by dye-labeled siRNAs, by monitoring the expression level of cellular eGFP using confocal analysis (79.6%, 80.4%, 76.4%, and 90.2% knockdowns resulted from silencing effects of non-, both-, sense-, and antisense-labeled siRNAs, respectively), as summarized in Table 1. Each of 3 dye-labeled siRNAs exhibited undiminished or even increased silencing effect for its target mRNA, which is by and large comparable with the non-labeled standard siRNA. These results reveal that the labeling of fluorescent dyes at the 5′ or 3′ end of siRNAs has a minimal effect on gene-silencing activity in HeLa cells as well, which is consistent with a previous report that RNAi functionality was not affected by permutations of label attachment sites in siRNA strands (Järve et al., 2007; Jagannath and Wood, 2009; Shin et al., 2011). The eGFP reporter gene-silencing assay in this study showed little impediment of functionality of 3 dye-labeled siRNAs, despite some dispute in the literature on the diminished efficiency of siRNAs caused by the end labeling (Holen et al., 2002; Ohrt et al., 2006), confirming that all of our constructs could retain the sufficient activities to justify their use as functionally active siRNAs in cells. It is also interesting to note that the gene-silencing functionality of the prepared siRNAs in HeLa cells is greater than that in HEK293T cells (Shin et al., 2011), although both cells are generally known to be highly siRNA transfectable.
Non-, both-, sense-, and antisense-labeled small interfering RNAs (siRNAs; 30 pmol each), respectively, were transfected into the enhanced green fluorescent protein (eGFP)-expressing HeLa cells, and confocal images were taken 24 hours after transfection, as shown in Supplementary Fig. S1. The expression levels of eGFP and the fluorescence intensities in cells were quantified.
Confocal image analysis of siRNAs labeled with FRET dyes for analysis of siRNA sustainability in live cells
In terms of imaging of siRNA labeled with FRET dyes, we transfected sense-labeled (or passenger-strand-labeled) siRNA into the eGFP-expressing HeLa cells that had been transfected with eGFP expression vector and grown for 24 hours, as previously reported (Shin et al., 2011). The confocal images for the dissociation patterns of siRNAs in HeLa cells are markedly different from in HEK293T cells (Fig. 2). On examining the fluorescence emission from cells after 0.5-hour, 1.5-hour, and 2.5-hour incubations (Fig. 2, middle), we detected both the donor (TAMRA at ∼581 nm) and acceptor (Cy5 at ∼667 nm) signals, arising from donor excitation (543 nm) and from acceptor excitation (643 nm), respectively, which indicates that both the donor and acceptor fluorophores are existent in the cells regardless of their connectivity. To evaluate the relative FRET signals in cells, the ratios between the fluorescence intensities at 581 nm and at 667 nm after 543-nm excitation were calculated as reported in the literature (Järve et al., 2007; Jagannath and Wood, 2009; Shin et al., 2011). Interestingly, we observed almost no FRET signal from the sense-labeled siRNA-transfected cells after 3.5-hour incubation (Fig. 2), demonstrating that the FRET dyes were gradually dissociated (i.e., degraded) and that both the fluorescence intensities of the dyes might be almost completely reduced probably by the efflux of the released TAMRA and Cy5. It should be noted here that the crosstalk between TAMRA and Cy5 needs to be considered when IFRET/ITAMRA is smaller than 0.08
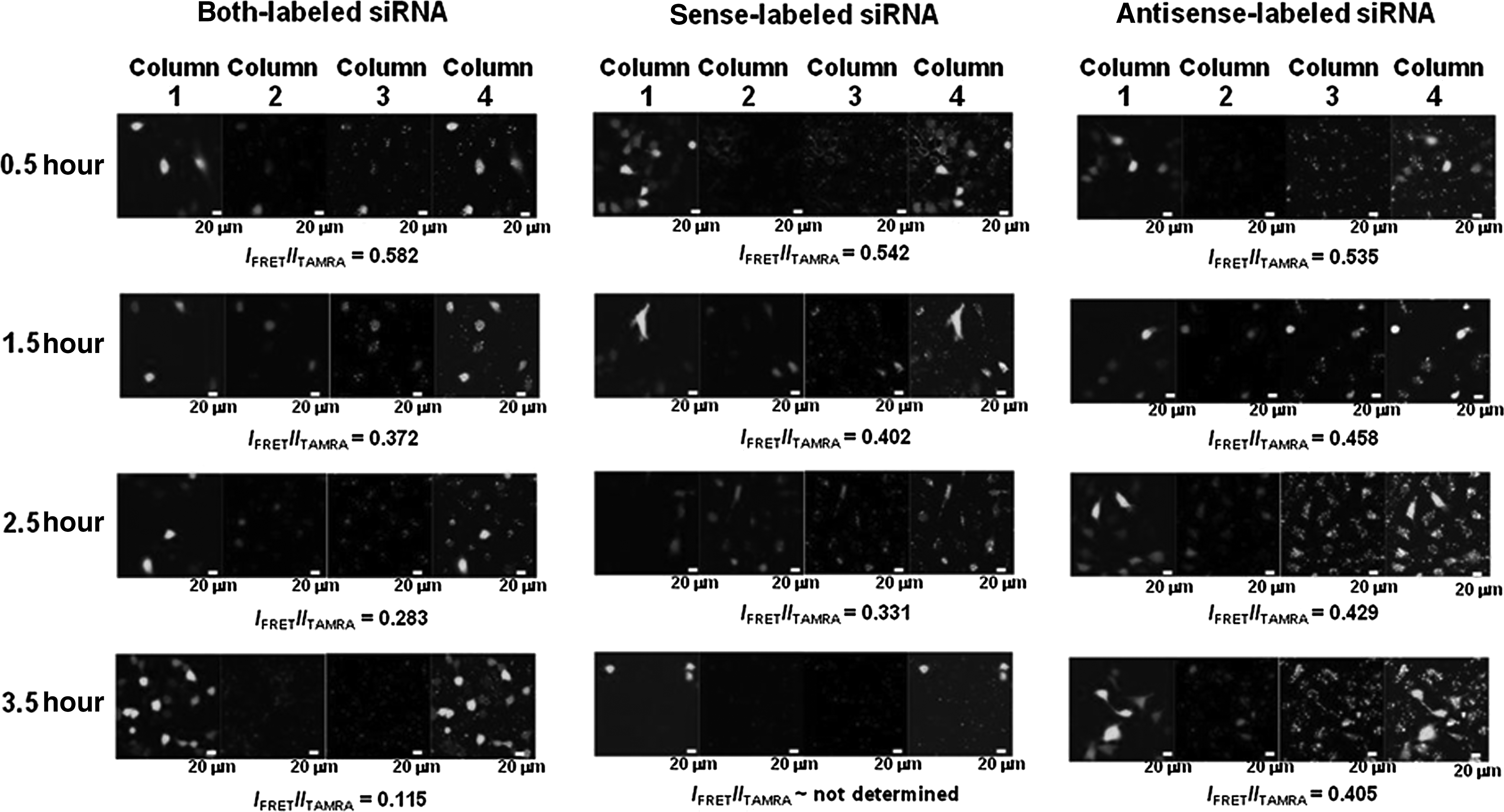
Confocal image analysis of siRNAs labeled with FRET dyes, showing fluorescence emission after 0.5-hour, 1.5-hour, 2.5-hour, and 3.5-hour incubation, respectively, from the enhanced green fluorescent protein (eGFP)-expressing HeLa cells into which both-, sense-, and antisense-labeled siRNAs were transfected, respectively. (Column 1, eGFP expression images at 509 nm after 488-nm excitation; Column 2, fluorescence from TAMRA at 581 nm after 543-nm excitation; Column 3, FRET images due to the energy transfer from TAMRA to Cy5 fluorescence at 667 nm after 543-nm excitation; Column 4, merged images). To evaluate the FRET signals in cells compared with the TAMRA emission intensities in cells, the ratios between the fluorescence intensities in Column 3 (IFRET=Itotal−Ibackground) and in Column 2 (ITAMRA=Itotal−I background) were calculated as reported in the literature (Järve et al., 2007; Jagannath and Wood, 2009; Shin et al., 2011).
We then transfected both-labeled siRNAs into the eGFP-expressing HeLa cells in order to monitor the amount of double-stranded siRNA in the cells over time and observed a strong FRET signal from the siRNA-transfected cells (Fig. 2, left). These results indicate that the 2 fluorescent probes were associated together in proximity and that the FRET signal we captured might be from double-stranded siRNAs. Importantly, we found that the fluorescence signal ratios reduced over time with the highest at 0.5 hours after transfection, which is a little faster than that of sense-labeled siRNAs. This comparison of the FRET signals as a function of time (0.582, 0.372, 0.283, and 0.115 after 0.5-hour, 1.5-hour, 2.5-hour, and 3.5-hour incubation, respectively) suggests that the dissociation (or melting) of the siRNA duplex occurs after the highest accumulation of siRNAs in cells, since the higher ratio represents the higher amount of the FRET pair in proximity in cells. The data also reveal that there would be no gene-silencing effect in the cells without any intracellular TAMRA's or Cy5's fluorescence resulting from successful siRNA transfection.
With the FRET pair, we selectively labeled passenger (or sense) strand and guide (or antisense) strand, respectively, of siRNAs against eGFP and followed the intracellular localizations of the dyes over time by confocal microscopy (Fig. 2, middle and right), indicating slightly different patterns, especially of Cy5. The confocal data also allowed us to follow the cleavage and/or degradation of the full-length RNA into shorter RNA products, because the cleavage and/or degradation of the strand with the FRET pair over time might lead to the decrease in the FRET signals (Shin et al., 2011). Figure 2 shows that the fluorescence signal ratios are 0.582, 0.372, 0.283, and 0.115 for sense-labeled siRNA after 0.5-hour, 1.5-hour, 2.5-hour, and 2.5-hour incubation (Fig. 2, middle), and 0.535, 0.458, 0.429, 0.405, and 0.324 for antisense-labeled siRNA after 0.5-hour, 1.5-hour, 2.5-hour, 3.5-hour, and 24-hour incubation (Fig. 2, right, and Supplementary Fig. S2), respectively. In consideration of the similar efficacy of gene silencing by the 2 dye-labeled siRNAs, these results strongly indicate that the antisense strand of siRNA is still intact even after 24-hour incubation and that cleavage/degradation of the passenger (or sense) strands of our siRNAs is particularly substantial in cells after transfection, which may explain why the gene-silencing efficiency of siRNAs against eGFP in HeLa cells is higher than that in HEK293T cells. Importantly, this result is consistent with the previous report on the higher transcript levels of Argonaute2, the only catalytic endonuclease among the Argonaute subfamily, in HeLa cells than in HEK293T cells (Meister et al., 2004), since a functional RNA-induced silencing complex (RISC) is known to assemble only around Agonaute2. This also strongly suggests that the sustainability of antisense strand in the cells should be taken into account to improve the transfection efficiency of siRNAs, while the effect of the transfection carrier on the transfection efficiency of siRNAs should also be considered. Figure 3 clearly shows the exponential decrease of the IFRET/ITAMRA values of the dye-labeled siRNAs in HeLa cells, which is intrinsically different from the in vitro stability of siRNAs, as demonstrated in Supplementary Fig. S3. Our results that the antisense strand which has the longer lifetime than the sense strand are supposed to be degraded under intracellular conditions of target-expressed cells need to be compared with the previous paper describing that siRNAs in cells persist intact for extended periods of time (Bartlett and Davis, 2007).
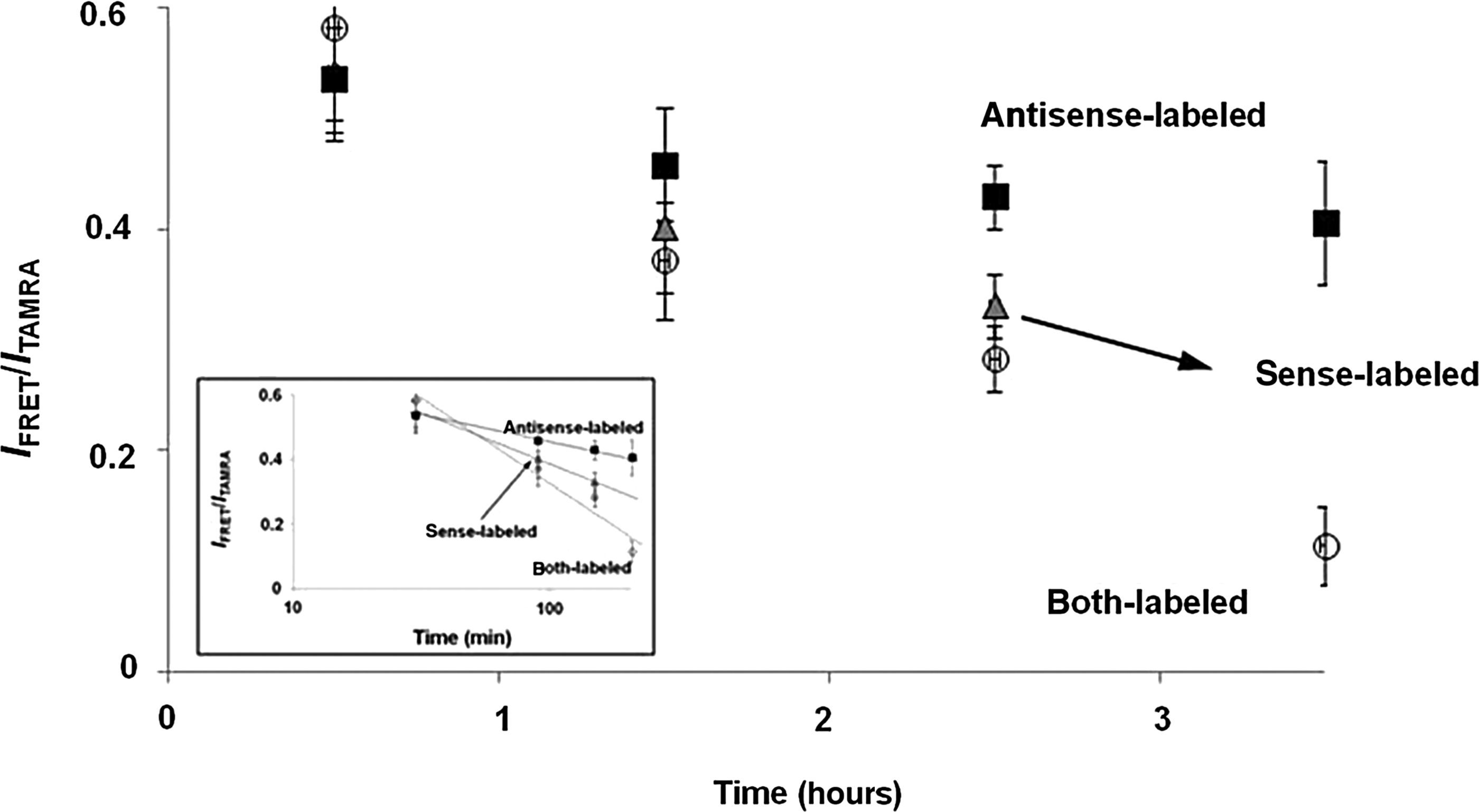
IFRET/ITAMRA values of dye-labeled siRNAs in HeLa cells as a function of incubation time. Experiments were performed in triplicate. The inset shows a logarithmic scale graph for the obtained data, demonstrating the exponential decrease of IFRET/ITAMRA values.
Single-molecule FRET study to track the fluorescence intensities of siRNAs with FRET dyes at the single-molecule level
Prompted by the results from the cell-based imaging experiments, we made use of a TIR-based single-molecule fluorescence resonance energy transfer (smFRET) method (Roy et al., 2008; Kim et al., 2012a, 2012b) in order to investigate the single-molecule spectroscopic events of siRNAs in HeLa cells, which might provide us with valuable information on its time-resolved dynamics of siRNAs in live cells. Single-molecule experiments, which can directly visualize and track individual siRNA molecules, are powerful tools in probing photophysical events because they reveal information hidden by ensemble averaging in bulk experiments, including static and dynamic heterogeneity, and because single-molecule sensitivity can allow for the detection of events too rare to perturb the ensemble-averaged signal (Roy et al., 2008).
We recently reported that the TIR-based smFRET method could be used to investigate the single-molecule spectroscopic events of quantum dot fluorescence induced by NADPH-dependent biocatalyzed transformation (Kim et al., 2012a). This method has been employed in the present study for imaging single siRNA molecules. To study the fluorescence properties of the single FRET-based siRNAs, our system is constructed around a commercial inverted microscope equipped with 532-nm diode-pumped solid-state laser system (Nd:YAG continuous crystal laser) and electron-multiplying charge-coupled-device (CCD) camera. The fluorescence signals from TAMRA and Cy5 that were collected by a water-immersion (prism-type) objective lens went through a long-pass filter to block out laser-light scattering. Signals lower and higher than 611 nm were separated by a dichroic mirror and detected by a CCD camera with up to 0.2-second time resolution. The observation area was 25 μm×50 μm.
Taking advantage of the fixed locations of siRNA-transfected cells on the chamber slide, we tracked the fluorescence intensities of the single siRNAs with a TIR-based smFRET instrument as a function of time (Fig. 4). Time-trace data of the fluorescence emission from each fluorophore were recorded under continuous 532-nm laser excitation for 20 seconds with a resolution of 0.2 seconds per scan, and it was found that the single-molecule spectroscopic events of the fluorescence changes of TAMRA and Cy5 in cells dramatically differed under constant laser excitation and that the fluorescence of TAMRA both from both- and sense-labeled siRNAs was gradually intensified over 40 minutes, strongly suggesting that the time-dependent FRET-mediated fluorescence changes at the single-molecule level could be caused by the dissociation-induced distance changes of siRNAs in live cells. Because single-molecule tracking can reveal the range of behaviors of the individual molecules and define characteristics of the cellular matrix in which they travel, methods for the detection of individual siRNA molecules using FRET-based probes are expected to provide the only means of seeing where these siRNAs go and how they get there.

Single-molecule images of HeLa cells transfected with dye-labeled siRNAs. For a chamber slide preparation, trypsinized HeLa cells were incubated at 37°C with lipofectamine 2000 and siRNAs for transfection and deposition on the naked glass surface for 30 minutes. After washing the surface with phosphate-buffered saline, the chamber slide was assembled with a coverslip. For the single-molecule fluorescence detection using the total internal reflection–based single-molecule FRET instrument, the chamber slide was mounted on the inverted microscope, after additional incubation for the indicated time under normal culture conditions. The resulting fluorescence signals were recorded in real time with a resolution of 0.2 seconds per scan for 40 seconds. The image is split into TAMRA (below 611 nm) and Cy5 (above 611 nm) emission channels, each 25 mm×50 mm.
Our observations are collectively consistent with the previous papers (FILIPOWICZ, 2005; Matranga et al., 2005; Miyoshi et al., 2005; SONTHEIMER, 2005; Leuschner et al., 2006; Shin et al., 2011), which report that double-stranded siRNAs specifically interact inside the cell with a number of proteins including the endoribonuclease Dicer that cleaves them from long precursor RNAs and steers incorporation of this product into a multiprotein complex referred to as the RNA-induced silencing complex (RISC). One strand of the siRNA duplex is retained in RISC and acts as a template for the sequence-specific identification and site-specific cleavage of mRNAs and concomitant reduction in gene expression, as summarized in Fig. 5.

Schematics for assembly of siRNA with RNA-induced silencing complex (RISC) and intracellular fate of each siRNA strand in cells. This schematic emphasizes our findings that indicate transfer of duplex siRNA to Argonaute2, the only catalytic endonuclease among the Argonaute subfamily, with either concurrent or immediate Argonaute-mediated cleavage of the passenger strand. Mature RISC includes only the antisense (or guide) strand of siRNA, which may increase the cellular sustainability of the antisense strand of siRNA. More details of the models can be found in the literature (Filipowicz, 2005; Matranga et al., 2005; Miyoshi et al., 2005; Sontheimer, 2005; Leuschner et al., 2006; Shin et al., 2011). With regard to this work, it should be noted that the transcript level of Argonaute2 in HeLa cells is reported to be higher than in HEK293T cells (Meister et al., 2004), which may explain why the gene silencing efficiency of siRNAs against eGFP in HeLa cells is higher than that in HEK293T cells.
Conclusion
To summarize, we report on the use of an advanced fluorescence-based method to probe the intracellular fate of siRNAs following their delivery into HeLa cells. The FRET probes were specifically designed to observe intracellular dissociation and decay of short synthetic RNAs in real time, thus providing the desired kinetic information in cells, and cell-based measurements demonstrated that the FRET signals due to the energy transfer from donor (TAMRA) to acceptor (Cy5) fluorescence could be used to study the dissociation/degradation of the labeled siRNA strands upon donor excitation. Intracellular FRET analysis revealed the asymmetric degradation as well as the time-dependent dissociation of each siRNA strand during the measured time period. These data underline the high intrinsic nuclease resistance of duplex siRNAs and prove that cellular persistence is much more critical for the single-stranded structure. Additionally, this study shows the potential of the delineated fluorescence-based technique for future research on biological behavior of siRNAs even at the single-molecule level. The fluorescence-based method presented here is a straightforward technique to gain direct information on siRNA integrity inside living cells and provides a bright outlook to learn more about the intracellular fate of siRNA therapeutics.
Footnotes
Acknowledgments
We are grateful to Prof. Young Dong Kim (Kyung Hee University) for his help. This work was supported by the National Research Foundation of Korea grant and the Korea Science and Engineering Foundation grant funded by the Korea Government (2009-0071058, 2011-0016385, 2011-0021956, and 2012-001680).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.