
Abstract
New natural and chemically modified DNA aptamers that inhibit HIV-1 activity at submicromolar concentrations (presumably via preventing viral entry into target cells) are reported. The new DNA aptamers were developed based on known intramolecular G-quadruplexes (G4s) that were functionally unrelated to HIV inhibition [the thrombin-binding aptamer and the fragment of the human oncogene promoter (Bcl2)]. The majority of previously described DNA inhibitors of HIV infection adopt intermolecular structures, and thus their folding variability represents an obvious disadvantage. Intramolecular architectures refold correctly after denaturation and are generally easier to handle. However, whether the G4 topology or other factors account for the anti-HIV activity of our aptamers is unknown. The impact of chemical modification (thiophosphoryl internucleotide linkages) on aptamer activity is discussed. The exact secondary structures of the active compounds and further elucidation of their mechanisms of action hopefully will be the subjects of future studies.
Introduction
G- (1) aptamers to reverse transcriptase (RT) (eg, the intramolecular antiparallel G4-forming ON d(GGGGG TGGGAGGGTAGGCCTTAGGTTTCTGA) [5]); (2) aptamers to the envelope glycoproteins gp120 and/or gp41 (eg, the tetramolecular parallel G4-forming thiophosphoryl ON d(TTGGGTT) [6] and terminally modified hexamer d(TGGGAG) [7,8]); (3) aptamers to the HIV integrase (IN) (eg, the ON d(GG GGTGGGAGGAGGGT) known as 93del, which adopts a dimeric “interlocked” parallel G4 structure with an AG4 pentad sandwiched between G-tetrads [9]; the thiophosphoryl ON d(GGGTTTTGGG), which folds into a dimeric antiparallel G4 [10]; the intramolecular antiparallel G4-forming ON Zintevir (also known as AR177 and T30177) d(G*TGGTGG GTGGGTGGG*T), where * is a thiophosphoryl internucleotide linkage [11]; and the Zintevir analogs d(GTGGTGGGTGGGTGGGT) (T30175), d(G*GGT GGGTGGGTGGG*T) (T30695), and d(GGGT)4 (T30923) [11–13] with ambiguous folding [14,15]).
Notably, some of these aptamers can act as multimodal HIV inhibitors. The “interlocked” aptamer 93del was initially selected against the RNase H domain of HIV-1 RT but exhibited anti-IN activity in vitro [16] and interfered with viral entry, reverse transcription, and integration in cells [17]. Similarly, Zintevir and its derivatives were initially selected against HIV-1 IN, but subsequent studies revealed that gp120 might be their primary target [18]. Aptamer binding to gp120 generally interferes with interactions between gp120 and the cellular receptors (CD4 and co-receptors), thereby preventing viral attachment.
Structure-activity and stability-activity relationships have been investigated to some extent for several G4 HIV inhibitors [19]. The most rigorously studied aptamers are probably the 5′-substituted Hotoda's ONs d(TGGGAG) [7,8], which target HIV-1 entry through gp120 binding [20]. These hexamers adopt a tetramolecular parallel G4 topology with four G-tetrads and one internal A-tetrad [21]. 5′-Terminal hydrophobic moieties (dimethoxytrityl, DMTr [22], 3,4-dibenzyloxybenzyl [20], tert-butyldiphenylsilyl [23] or other aromatic groups [24]) significantly contribute to aptamer activity, presumably by direct participation in aptamer-protein interactions [25]. It has been also been assumed that the modifications facilitate G4-folding, but recent findings argue against this assumption [26].
Other modifications that enhance aptamer activity include the introduction of a 2-hydroxy-ethylphosphate group at the 3′-terminus [7,8] and conjugation with monosaccharide (glucose or mannose) residues [27]. Substitutions of the core guanines for conformationally constrained “locked” nucleic acid monomers at certain positions resulted in potent aptamers with increased thermal stability [28]. However, the correlation between G4 stability and overall aptamer activity is not strict. Bulky pyrene derivatives at the 5′-terminus alter G4-folding and lead to activity loss [28], which argues for the importance of parallel G4-folding for efficient target recognition.
To summarize, the analysis of active G4-DNA HIV inhibitors reveals the following tendencies:
• The most common scaffolds are the antiparallel G4 core and the four-tetrad parallel G4 core. • G-T-rich sequences are abundant, which is hardly surprising because thymine in loops is thermodynamically more favorable than adenine (its exposure to solution results in lower solvation entropy), whereas C-rich fragments (if present) induce the formation of Watson-Crick hairpins. Interestingly, adenines in G4-forming sequences may also be beneficial (eg, participate in the formation of noncanonical tetrads or pentads). • Terminal chemical modifications (especially hydrophobic modifications) can increase G4 thermal stability, whereas local backbone modifications, such as thiophosphoryl internucleotide fragments, are typically used to endow aptamers with resistance to nucleases and to enhance aptamer-binding affinity [29].
Many potent inhibitors of HIV-1 entry (eg, d(TTGGGTT) [6], Hotoda's aptamer and its derivatives) form intermolecular structures. The intermolecular G4 arrangement is generally prone to conformational polymorphisms and concentration-dependant rearrangements, which complicates handling of these aptamers and poses limitations on their practical application. Several attempts have been made to overcome this problem in Hotoda's hexanucleotides. Piccialli and colleagues employed the TEL-ODN strategy (tetra-end-linked oligodeoxynucleotides) [30,31] (ie, they joined the 3′-termini of the four 5′-end-substituted hexanucleotides through a tetra-branched linker to obtain active pseudointrameric G4s). Although this strategy has proven to be rather efficient, the preparation of TEL-G4 requires significant synthetic effort and we have chosen a different approach here. We designed a series of native and chemically modified ONs using intramolecular G4s as basic scaffolds and tested their anti-HIV activities.
The abovementioned multimodal function of several HIV inhibitors and the diverse cross-reactivity of genomic G4s prompted us to search for appropriate G4 scaffolds among already known but seemingly unrelated intramolecular aptamers. SELEX, which is the gold standard methodology for the development of specific aptamers, is often time-consuming and suffers from a relatively low success rate [32]. We hypothesized that exploiting the potential of the existing aptamer pool might be as productive as de novo selection if preliminary knowledge about the structure-activity relationship was available.
The thrombin-binding aptamer (TBA) [33] has been comprehensively characterized and is arguably one of the most celebrated G4 structures. TBA forms an antiparallel chair-type 2-tetrad G4 with Tm = 52°C ± 1°C in the presence of 100 mM KCl [34]. The GT-richness and the antiparallel folding of this aptamer suggested that it could possess some antiviral activity (see the general features of known active G4-DNA HIV inhibitors). TBA has not been considered as an antiviral agent to date but has been shown to interact with the HIV nucleocapsid protein [35].
TBA has also demonstrated significant antiproliferative activity in human cell cultures [36]. Until recently, there had been no follow-up on the latter finding because the anticoagulant properties of TBA hampered its potential anticancer applications. Eventually, a suitable chemical modification that preserved the antiproliferative activity of TBA over its anticoagulant actions was reported [37]. This finding is likely to reinvigorate interest in TBA analogs with respect to cancer treatment and illustrates the multifunctional potential of aptamer scaffolds.
In the context of the transmission of viral infection, blood clotting inhibition may be a double-edged sword. For catheter-related bloodstream infections, the dual antiviral-anticoagulant action of the aptamer may be an advantage because it will simultaneously solve the problem of catheter blockage [38]. However, in cases of vaginal transmission, anticoagulants might exacerbate the effects of ulceration. Some polyanionic microbicides that have entered clinical trials have demonstrated anticoagulant activities [39,40]; therefore, the risk of ulceration is a common problem.
Materials and Methods
ON synthesis, purification, and MS analysis
ONs were synthesized on a Biosset ASM-800 DNA synthesizer following standard phosphoramidite protocols and purified by preparative scale reverse phase high-performance liquid chromatography (HPLC) on a 250 mm × 4.0 mm Hypersil C18 column with detection at 260 nm and a 12%–24% gradient of CH3CN in 0.1 M ammonium acetate buffer. The DMTr protection group was removed via treatment with 80% acetic acid for 20 min (this step was skipped for DMT-thio-BclON). The detritylated ONs were further purified in a 0%–12% CH3CN gradient in 0.1 M ammonium acetate buffer. The purity of all ONs was determined to be 95% by HPLC. The matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra of the ONs were acquired on a Bruker Microflex mass spectrometer in linear mode (20 kV). Each spectrum was accumulated using 200 laser shots (N2 gas laser, 337 nm). A solution of 35 g/mL of 3-hydroxypicolinic acid with dibasic ammonium citrate was used as the matrix.
Circular dichroism, ultraviolet-melting and fluorescence resonance energy transfer experiments
ONs were dissolved in a 20 mM Tris-HCl buffer (pH 7.5) containing 10 mM KCl, 100 mM KCl or 100 mM LiCl, denatured at 95°C for 5 min and snap cooled on ice before all measurements to ensure intramolecular folding. The ultraviolet (UV) spectra, circular dichroism (CD) spectra and UV melting curves were recorded on a Chirascan spectrophotometer (Applied Photophysics) equipped with a thermostated cuvette holder. The melting temperatures of the quadruplexes were estimated from the maximum in the first derivative of the melting curve. The absorbance was registered every 1°C across the 20°C–90°C temperature range (heating rate = 0.5°C/min). The rotational relaxation times (RRTs) of ethidium bromide (EtBr) bound to the ONs were determined as described in Ref. [41]. In the fluorescence resonance energy transfer (FRET) experiments, the 6-carboxyfluorescein (FAM) fluorescence was measured with a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies) at 20°C. The excitation wavelength was 480 nm. The polarization of FAM fluorescence at 520 nm was determined as described in Ref. [41].
Assessment of ON inhibitory activity
The Jurkat cell line was cultured in RPMI 1640 medium containing 20% fetal calf serum, 4 mM
Toxicity studies
The influence of ONs on cell proliferation was measured on suspension Jurkat cells using MTT Cell Proliferation Kit (OZ Biosciences). Cells were cultured in 96-well plates. ONs were added to culture medium at concentrations of 0, 0.1, 1, 5, 10, and 20 μg/mL for 48 h. Each concentration was evaluated in triplicate. The metabolic activity (farmazan production) was measured according to the suspension cells protocol provided by the manufacturer. The absorbance of the converted dye was measured on a plate reader at 570 nm and a reference wavelength at 650 nm. The signal was calculated as OD570–OD650, and the results were expressed as a percentage of control (nontreated) cells.
The influence of ONs on cell viability was measured on Jurkat cells by fluorescence-activated cell sorting (FACS) analysis after propidium iodide (PI) staining of unfixed cells. Cells were cultured in 96-well plates. ONs were added into culture medium at concentrations of 0, 0.1, 1, 5, 10, and 20 μg/mL for 48 h. After treatment PI staining solution was added to each sample just before FACS analysis.
Photonic crystal surface wave experiments
Streptavidin-coated 1D photonic crystal slides were prepared as described in Ref. [44]. The surface was blocked with a bovine serum albumin (BSA) solution in phosphate-buffered saline (PBS) buffer [10 mM NaH2PO4/Na2HPO4 (pH 7.4), 137 mM NaCl, and 2.7 mM KCl]. The biotinylated ON solution (50 nM) in PBS was pumped through the working chamber until signal saturation was achieved and washed with PBS. The gp120 solution (various concentrations) in PBS was subsequently injected and run over the ON-coated 1D PC surface for ∼1,000 s (time required for signal saturation); then, the chamber was washed with the buffer for an additional 500 s (flow rate = 50 μL/min).
Streptavidin was purchased from Sigma-Aldrich, gp120 was purchased from Speed BioSystems, and BSA was purchased from Thermo Fisher Scientific.
Molecular modeling
Model preparation for the docking experiments
Three-dimensional models of the aptamers were obtained using the nuclear magnetic resonance -based structure of unmodified TBA in complex with human thrombin PDB 1HAO [45]. The protein was removed, and in the case of F-thio-TBA one oxygen atom of each phosphate internucleotide linkage in TBA was substituted with sulfur to produce diastereomers with all-R, all-S, or alternating R/S P configurations. Partial atomic charges of the thiophosphoryl linkage atoms were obtained by single-point energy quantum mechanics calculations. Electron density in thiophosphoryl linkages (including C3′ and C5′) was calculated by the ab initio quantum mechanical method (density functional theory [46]) with hybrid exchange-correlation functional B3LYP and the 6-31G(d) basis set. Based on the calculated electron density distribution, the partial atomic charges were computed using the Mulliken's population analysis scheme [47]. The choice of the scheme for calculating partial atomic charges is explained in [44]. All quantum mechanics simulations were carried out using the Gaussian 09 program.
The gp120 model was taken from PDB 2B4C [48] and optimized using the SYBYL 8.0 molecular graphics software package (Tripos, Inc.). Unfavorable van der Waals interactions were removed by short molecular mechanics minimizations using Powell's method [49] with the following settings: estimation of the partial atomic charge with application to Gasteiger–Hückel method [50], assignment of the atom types from TRIPOS force field [51], nonbonded cutoff distance equal to 8 Å, solvent effect accounting via the distance-dependent dielectric function, 1,000 iterations, simplex method in the initial optimization, and energy gradient convergence criterion with a threshold of 0.05 kcal mol−1 Å−1.
Docking
Flexible docking of the aptamers to full-sized gp120 was performed with Autodock 4.2 [52] to define the putative binding sites. The protein and aptamers were prepared for docking using AutodockTools (ADT Version 1.5.4). The 3D grid maps (126 × 126 × 126 points with 0.375 Å spacing) for each atom type were computed using AutoGrid4. Additionally, electrostatic and desolvation maps were calculated. Because viral adsorption and fusion occur via gp120 interactions with the CD4 receptor and chemokine receptor CCR5 with active participation of the V3 loop, docking was performed at areas containing the CD4- and CCR5-binding sites in gp120 and its V3 loop.
The side chains of the amino acid residues LYS97, ASN279, LYS282, HIS363, ASN461, ARG469, ARG476, and ARG480 were flexible upon aptamer docking to gp120 in the proximity of the CD4-binding site. The side chains of GLN301, ARG304, LYS305, ARG315, ARG327, HIS308, ARG419, GLN422, and LYS432 were flexible upon docking near the CCR5-binding site. The side chains of THR297, ARG298, ASN300, GLN301, ASN302, ARG304, LYS305, THR303, ILE307, HIS308, ARG315, ARG327, ASP325, PHE317, TYR318, HIS330, SER306, and ILE309 were flexible upon docking to the V3 loop. The G4 core was fixed in the aptamers but the nucleobases in the loops were allowed to rotate around the glycosidic bonds.
The Lamarckian genetic algorithm (GA-LS) [53], which is a hybrid of a genetic and local search algorithm, was applied to identify the most probable binding site. The following GA-LS parameters were used: 100 GA-LS runs, 2,500,000 energy maximum, maximum of 27,000 generations, and mutation and crossover rates of 2 and 0.8, respectively. The Pseudo-Solis and Wets parameters were used for the local search algorithm with the number of iterations set to 300. The starting positions and conformations of the aptamers were random. The torsion angle rotation step was 50°. After docking, all generated structures were clustered with a root-mean-square tolerance of 2 Å from the lowest energy. Free energies of aptamer binding with the gp120 sites were estimated using force field scoring functions and the formula:
The parameter ΔGelec was calculated by the distance-dependent dielectric of Mehler and Solmajer [54]. Van der Waals interactions (ΔGvdW) were estimated using the Lennard-Jones potential and atomic parameters from the AMBER force field. For scoring of the H-bond energy (ΔGH-bond), the 10/12 potential was used with a maximum well depth of 5 kcal/mol at 1.9 Å for hydrogen bonds with oxygen and nitrogen atoms and then multiplied by the function estimation rate from the ideal H-bonding geometry. To estimate the desolvation energy (ΔGdesolv), we used the atomic fragmental volume and solvation parameters derived from the method of Stouten et al. [55]. Each term was multiplied by semi-empirical weighting constants by fitting a training set of complexes with known structures to the binding affinity data. The scoring function in the docking procedure also involved the conformational entropy ΔGtors. The change in the aptamer conformational mobility was solely estimated by calculating the number of dihedral angles.
The precise calculation of ΔGtors might be difficult in our models for the following two reasons: (1) conformational changes in gp120 and the aptamers could substantially contribute to ΔGtors but could not be accurately estimated and (2) the weighting constant for ΔGtors might be incorrect because the regression multipliers for this parameter were obtained for chemical structures that were significantly different from the investigated aptamers. Therefore, the ΔGtors values were omitted from the analysis.
Molecular dynamics simulation
The explicit (TIP3P) solvent simulations were performed using the Amber 14 suite in a cubic water box with periodic boundary conditions imposed. The parameters of all atoms needed for interatomic energy calculations were taken from the force fields ff14SB and GLYCAM_06j-1. The negative charges were neutralized by adding Na+ ions, and ∼19,000 water molecules were employed for solvation. A K+ cation was placed in the center of the aptamer between the quartets to stabilize the structure. The structures were minimized before the simulations.
First, the locations of the solvent molecules were optimized for 1,000 steps (500 steps of a steepest descent minimization followed by 500 steps of a conjugate gradient minimization), with all the solute atoms being restrained to their positions with a force constant of 500 kcal mol−1 Å−2. Then, the complex structure was optimized without any restriction for 2,500 steps (1,000 steps of steepest decent followed by 1,500 steps of conjugate gradient). Subsequently, a gradual heating to 300 K over 20 ps was performed. To avoid wild fluctuations in the system at this stage, weak harmonic restraints with a force constant of 10 kcal mol−1 Å−2 were used for all atoms with the exception of the solvent atoms. All bonds to hydrogen atoms were constrained using the SHAKE algorithm [56], which allowed a time step of 2 fs. The nonbound one to four van der Waals and electrostatic interactions were scaled by standard amber values (SCEE = 1.2, SCNB = 2.0). The cutoff for van der Waals interactions was set to 10 Å. Long-range electrostatics was calculated using the particle mesh Ewald method.
The molecular dynamics (MD) simulations in a production phase were performed at p = 1 atm and T = 300 K under the control of a Langevin thermostat with a collision frequency of 1 ps−1. The trajectory length was 8 ns. Snapshot visualization was performed using VMD software (www.ks.uiuc.edu/Research/vmd). The molecular mechanics with the generalized Born and surface area solvation (MM-GBSA) analysis was performed as described in Ref. [44]. Ensemble averages and standard deviations of the free energy and its components were calculated by means of Gibbs–Boltzmann distribution for the ensemble of conformations obtained from the trajectory computed during the production phase.
Results and Discussion
ON design and secondary structure analysis
As has been mentioned in the Introduction section, TBA could be considered as a scaffold for developing anti-HIV candidates because of its relative structural resemblance to known HIV inhibitors, but the anticoagulant activity has to be minimized (eg, via chemical modification). Previously, we analyzed TBA analogs with thiophosphoryl internucleotide linkages [34,44,57]. The thio-modification was shown to reduce anticoagulant activity [57] and had various effects on the thermal stability of the aptamer (from moderate destabilization to slight stabilization) depending on the position and number of thio-linkages [34]. The modification is also known to increase toxicity (a major disadvantage) and improve ON resistance to nuclease cleavage [58]. We assumed that TBA analogs with high thermal and enzymatic stability but reduced anticoagulant activity might merit testing as microbicide candidates.
The sequences of all ONs tested are provided in Table 1. Thio-TBA is a TBA derivative with thiophosphoryl internucleotide linkages in the TT loops. It is a slightly less potent thrombin inhibitor than TBA but has slightly enhanced thermal stability (Tm = 54°C ± 1°C in the presence of 100 mM KCl) and substantially enhanced resistance to nuclease digestion [57]. F-thio-TBA (the “fully modified” aptamer) bears thiophosphoryl internucleotide linkages throughout the chain. It is less thermostable than TBA (Tm = 45°C ± 1°C in the presence of 100 mM KCl) but exhibits no anticoagulant activity [44]. Both thio-analogs of TBA retain the antiparallel folding pattern (see Ref. [34] for CD spectra and melting curves).
The DMT group is cleaved under the conditions of MALDI-TOF MS.
IC50 values of control non-ON HIV-1 inhibitors chitosan sulfate and dextran sulfate were 0.06 ± 0.03 and 0.11 ± 0.04 μg/mL, respectively. N25 is a mixture of random sequence 25-mers obtained by solid-phase synthesis using an equal mixture of all four phosphoramidites at each step. Its activity inhibitory was within experimental error.
Anti, antiparallel intramolecular G4; CD, circular dichroism; hybrid, hybrid (mostly parallel) intramolecular G4 (determined by CD and RRT analysis); MALDI-TOF MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; n/a, not analyzed; ON, oligonucleotide; RRT, rotational relaxation time; TBA, thrombin-binding aptamer; Thio, thiophosphoryl internucleotide linkage.
The other scaffold used in this study is a fragment of the human Bcl2 promoter region (chr18: −60985942 to −60985966, BclON) (Table 1). Direct involvement of this highly conserved G4 motif, located 42 nucleotides upstream of the translation start site, in post-translational modulation of Bcl2 expression has been demonstrated [59]. A number of genomic G4 motifs have been previously considered as aptamer candidates [60] or ON decoys [61,62], and a promoter-derived G4 aptamer selection method (G4PAS) has been proposed [63]. Generally, genomic noncanonical structures appear to be promising aptamer scaffolds. Parallel intramolecular G4s from oncogene promoters are probably most suitable for antitumor drug design because their folding is associated with cancer-selective cell binding and antiproliferative activities [64,65]. We hypothesized that these G4s might also have potential utility as antiviral agent scaffolds because parallel G4s are rather common among known ON inhibitors of HIV (see Introduction section).
To date, the BclON secondary structure has not been reported but its RNA analog has been shown to form an intramolecular parallel G4 [59]. The CD spectra of BclON and its thiophosphoryl derivative F-thio-BclON (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/nat) bear features that are characteristic of both parallel (a positive band near 265 nm and a negative band near 245 nm) and antiparallel G4s (a positive band near 295 nm). Thus, BclON and F-thio-BclON adopt hybrid topologies. The contribution of parallel folding (stacked G-tetrads with the same polarities) is more pronounced. In the presence of 100 mM KCl, the Tm values of BclON and F-thio-BclON were close to or above 80°C; the lower plateau of the melting curve was not observed and we were unable to accurately compare thermal stabilities. Therefore, we performed melting experiments (Supplementary Fig. S1B) at a moderate (10 mM) salt concentration (TmBclON = 58°C ± 1°C; TmF-thio-BclON = 62°C ± 1°C).
BclON-mut is a mutant of BclON designed to form no intramolecular G4 structures. Indeed, a negative peak at 295 nm was absent in the thermal difference spectrum of this mutant (Supplementary Fig. S1C) and no UV-melting at 295 or clear CD-melting at 265 nm was observed. The CD spectrum (Supplementary Fig. S1A) resembles a parallel quadruplex but the molar ellipticity at 265 nm is low, which argues against stable G4.
Intramolecular folding of BclON and BclON-mut after rapid annealing was confirmed by analyzing the RRTs of EtBr bound to ONs, which are roughly proportional to the hydrodynamic volumes of the complexes (Supplementary Fig. S1D). To clarify whether BclON-mut can adopt dimeric or tetrameric structures at higher concentrations, additional experiments with the slowly annealed samples were performed (Supplementary Fig. S2). Apparent concentration-dependence and salt-dependence of molar ellipticity at 265 nm and relatively sharp melting curves at 50 μM BclON-mut concentration indicate the presence of intermolecular G4s. At submicromolar concentrations their contribution would be low.
A traditional definition of a G4 motif that stipulates uninterrupted G3+ runs suggests three-tetrad BclON/F-thio-BclON structures. The ratio of CD amplitudes at 295 and 265 nm argues for two stacked quartets with similar polarities and a third quartet with the opposite polarity [66]. However, high thermal stabilities of these G4s suggest more complex structures with additional stacking (eg, four-tetrad G4s with G-vacancies [67] and/or bulging nucleotides [68]) (Supplementary Fig. S1E). The exact secondary structures of BclON and its derivatives hopefully will be addressed in future studies.
General assessment of anti-HIV-1 activity
The inhibitory activities of the ONs were assessed using a lentiviral system [42,43]. Jurkat cells were transduced with pseudo-HIV particles containing HIV enzymes, recombinant GFP-coding RNA, and the HIV gp160 surface proteins (gp41 and gp120). Cell transduction resulted in GFP expression. The transduction efficiency in the presence of the ON inhibitors was monitored by GFP fluorescence. The results are summarized in Fig. 1 and the IC50 values are given in Table 1. The IC50 values were calculated by fitting the experimental data (transduction efficiencies at different inhibitor concentrations) to the standard dose–response equation (Supplementary Fig. S3). Interestingly, the dose–response curves obtained for the TBA derivatives suggest Hill coefficient [Hill Slope (HS)] values very close to 1 (which argues for 1:1 binding), whereas in the case of the BclON series, HS values other than 1 were obtained (which indicates multiple ligand binding) [69].

Suppression of HIV-1 lentiviral transduction by G4 ONs and their derivatives. Lentiviral transduction was estimated by calculating the number of fluorescent cells transduced with pseudo-HIV particles in the presence of the ON inhibitors. Known non-ON inhibitors of viral entry (chitosan sulfate and dextran sulfate) were used as positive controls. N25 is a mixture of random-sequence 25-mer ONs that is used as the negative control. The data represent mean values of two experiments performed in duplicate. ONs, oligonucleotides.
To exclude the possibility of ON toxicity interference with transduction efficiency measurements, we evaluated ON effects on proliferation and viability of Jurkat cells using the standard MTT test and FACS analysis (Supplementary Fig. S4). No significant effects were observed at ON concentrations up to 20 μg/mL.
All ONs with the exception of TBA and thio-TBA demonstrated relatively high inhibitory activities. The BclON derivatives were generally more active than the TBA derivatives. Unmodified TBA demonstrated minimal activity (comparable to that of the random ON N25 used as a negative control), whereas unmodified BclON and BclON-mut inhibited lentiviral transduction in the nanomolar range.
The well-known intramolecular G4 inhibitor of HIV infection Zintevir (IC50 = 2.4 μg/mL according to Ref. [18]) has been reported to be less efficient than the control non-ON inhibitor dextran sulfate (IC50 = 0.8 μg/mL according to Ref. [18]). In our experiments, BclON-mut, F-thio-BclON, and DMT-thio-BclON (IC50 values: 0.08 ± 0.03, 0.040 ± 0.003, and 0.018 ± 0.002 μg/mL, respectively) were superior to dextran sulfate (IC50 = 0.11 ± 0.04 μg/mL, Fig. 1). Although direct comparison of our data with the data in the literature would be inappropriate because different methods, conditions, and cell lines were used, it is apparent that F-thio-BclON and DMT-thio-BclON are highly potent inhibitors of lentiviral transduction that should be tested against wild-type HIV and can be considered as microbicide candidates. Although BclON and BclON-mut are less active, they may be even more promising because lower (if any) general toxicity can be expected for these natural ONs relative to the thiophosphoryl ONs.
ON binding with the HIV gp120 glycoprotein
Many known G4 inhibitors of HIV-1, including aptamers with multimode action, have been reported to prevent viral entry and have demonstrated affinities toward the viral glycoproteins [6,18,23]. The HIV envelope glycoprotein consists of an exterior domain (gp120) and a transmembrane domain (gp41). Interactions with a cellular CD4 receptor (the attachment step of viral entry) trigger conformational rearrangements in gp120 that allow it to engage a co-receptor (ie, the chemokine receptor CCR5 or CXCR4). Co-receptor binding exposes a hydrophobic region (fusion peptide) in gp41and leads to the fusion of the viral envelope with the cellular membrane [70].
Our lentiviral transduction experiments were performed in the absence of transfectants. Therefore, we assumed that the observed inhibitory activities of BclON and the TBA derivatives could be at least partially attributed to their interference with pseudo-viral entry. Additionally, an ON interaction with intracellular targets cannot be excluded because ONs (especially thiophosphoryl ONs) can theoretically enter cells at pharmacologically relevant concentrations even without transfection reagents [71].
To verify the hypothesis of viral attachment inhibition, we studied interactions of the leading unmodified ONs BclON and BclON-mut with recombinant gp120 using a photonic crystal surface wave biosensor (http://pcbiosensors.com) with independent registration of the angle of total internal reflection from the liquid [72]. The first implementation of this method for an investigation of aptamer-protein binding was reported in our previous work [44]. Here, we synthesized biotinylated analogs of BclON and BclON-mut [d(GGGGGCCGTGGGGTGGGAGCTGGGGTTTTT)-biot (the T5-linker is in italics; m/z calc. [M+H]+, 9884; found, 9888) and d(GGGGGCCGTTTTTTGGGAGCTGGGGTTTTT)-biot (m/z calc. [M+H]+, 9784; found, 9785), respectively].
The biotinylated ONs were immobilized on the streptavidin-coated biosensor working surface and different gp120 concentrations in PBS buffer were added. The obtained sensorgrams are shown in Fig. 2A (top graphs). Equilibrium increments of the effective adlayer thickness upon gp120 binding with the immobilized ONs were used to assess the binding constants (Fig. 2A, bottom graphs). The ONs demonstrated comparable affinities toward gp120 (KDBclON = 86 ± 17 nM and KDBclON-mut = 143 ± 79 nM). Interestingly, the BclON/gp120 KD value was rather close to the BclON IC50 value, which indirectly supports the assumption that the anti-HIV activity of BclON is primarily due to its interaction with gp120.
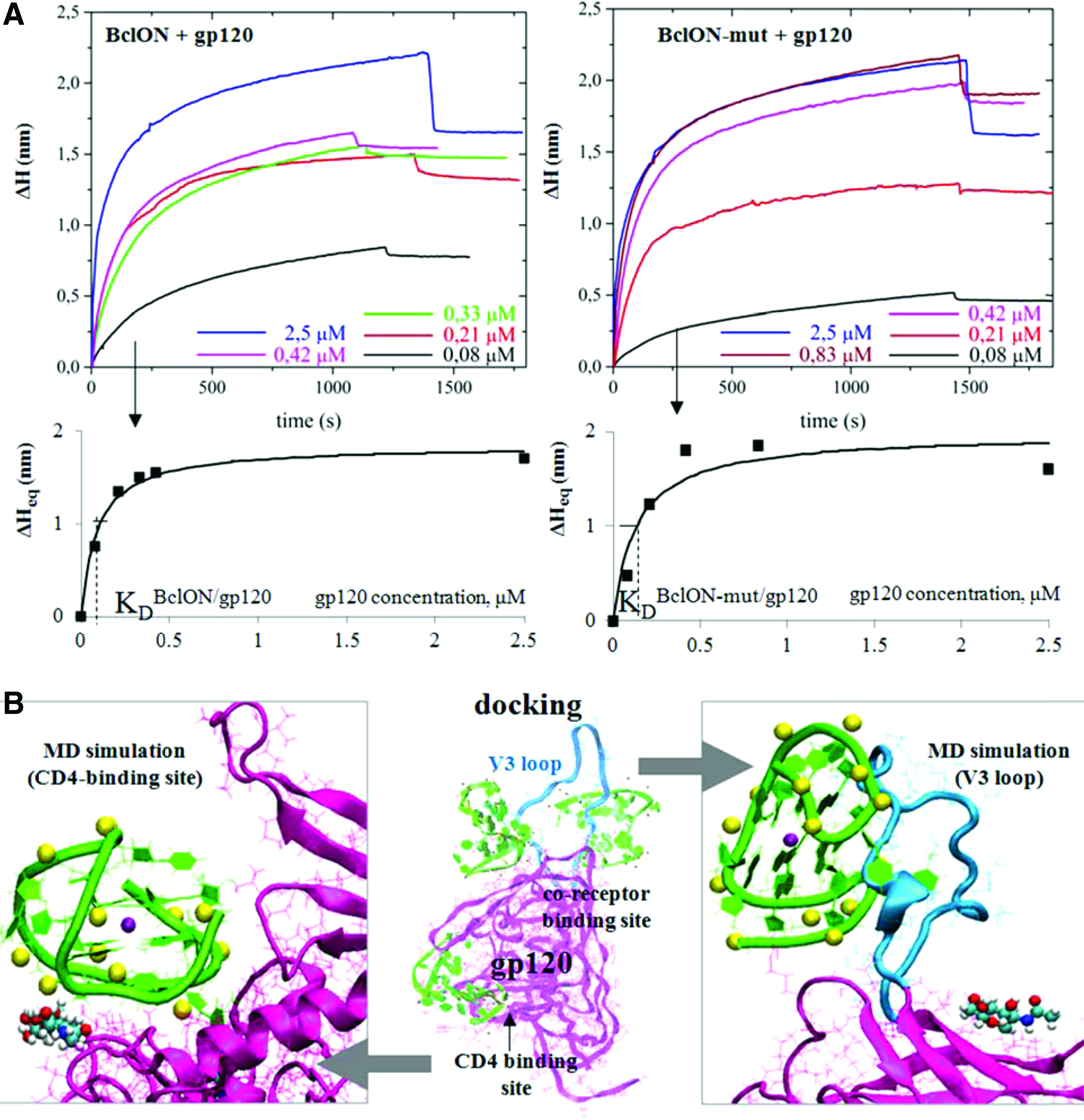
ON interactions with HIV-1 gp120.
To clarify whether BclON retained the G4 conformation or was unwound upon binding, we chose the FRET method (Supplementary Fig. S5), which allowed us to use low ON and protein concentrations (close to the IC50). We synthesized an analog of BclON bearing a fluorescent label (FAM) at the 5′ terminus and a respective quencher (BHQ1) at the 3′ terminus (FAM-BclON-BHQ, m/z calc. [M+H]+, 9050; found, 9052). In the presence of KCl or NaCl, this analog adopts a G4 conformation, FAM and BHQ are drawn together, and the FAM fluorescence is partially quenched. G4 unwinding (ie, upon heating and in the presence of LiCl) results in a ∼2-fold increase in the fluorescence intensity (Supplementary Fig. S5A).
We monitored changes in the FAM fluorescence intensity upon the addition of gp120 or pseudo-HIV virions (Supplementary Fig. S5B) in Na+/K+-containing working buffers at 20°C (G4 was initially folded under these conditions). The final pseudo-HIV concentration was equal to the concentration used in the activity tests. Fluorescence polarization was also measured in the experiments with gp120.
As expected, the fluorescence polarization significantly increased upon binding (Supplementary Fig. S5B, the right panel). The increase in the fluorescence intensity (both in the experiments with gp120 in PBS and the experiments with pseudo-HIV in the culture media) was relatively small compared with the increase induced by G4 unwinding. The gp120-bound probe would be at least partially shielded from water, so the observed small fluorescence enhancement may be attributed to FAM relocation into a more apolar environment (similar effects have been reported previously for other probes, including fluorescent TBA analogs [73]). Thus, our FRET data indicate that FAM-BclON-BHQ is likely folded in complexes with gp120 and pseudo-HIV. Of course, these results cannot be considered conclusive evidence for the maintenance of G4 in BclON because the label and the quencher may interfere with the binding.
The impact of chemical modifications on aptamer activity
As evident from the IC50 data in Table 1, the thiophosphoryl modification enhanced the anti-HIV activities of the tested ONs. Even partial modification (introduction of thiophosphoryl linkages into TT loops) imparted substantial activity to the initially inactive TBA. Modification throughout the chain was beneficial in both the TBA and BclON series. The addition of a terminal hydrophobic group (DMTr) further increased the F-thio-BclON activity. The effect of the DMTr group was in agreement with previously reported data for Hotoda's G4 ONs that inhibited gp120 binding to CD4 [22].
The positive effect of thio-modification on activity has been observed for a number of anti-HIV aptamers [6,11]. This effect may be due to extra-specific interactions with the target protein. Another possible explanation is that thio-ONs act similarly to dextran sulfate, cyclodextrin sulfate, and heparin (ie, by blocking nonspecific attachment of HIV virions to the cell surface, which precedes the gp120/CD4 interactions) [74].
Thio-modification enhances ON affinities for many proteins due to increased hydrophobicity, and high sequence-independent antiviral activity has been reported for fully modified thiophosphoryl ONs and their 2′-O-methylated derivatives [75]. We hypothesized that our thio-aptamers interacted with both viral glycoproteins and cell surface proteins. The latter type of interaction has not been properly analyzed and hopefully will be the subject of future investigations. Here, we report a preliminary estimation of the contribution of the thio modification to specific target recognition based on molecular modeling experiments (docking of the almost inactive TBA and its active F-thio-analog to gp120 and MD simulations for the aptamer/gp120 complexes).
Aptamer docking to gp120 (PDB: 2B4C) was performed to identify the most likely binding sites. Because F-thio-TBA exists as a mixture of a large number of diastereomers (214) with different P configurations, we were unable to model them all; therefore, we only considered three “boundary” diastereomers: (R)-F-thio-TBA (all P atoms in the R configuration), (S)-F-thio-TBA (all P atoms in the S configuration), and (R/S)-F-thio-TBA with alternating R and S configurations. Figure 2B displays the results of the (R/S)-F-thio-TBA docking. For the other diastereomers, see Supplementary Fig. S6. The free energies of aptamer binding with gp120 in the CD4-binding site, the co-receptor-binding site, and the V3 loop estimated using Autodock scoring functions were almost similar (Supplementary Table S1). The aptamers tended to interact with the protein via TT loops.
To further investigate the possible binding modes, we performed MD simulations of the aptamer complexes with the CD4-binding site and the V3 loop. The starting positions and conformations of the aptamers were determined from the docking experiments. In most of the complexes with the co-receptor-binding site, the V3 loop was also involved, so we considered such complexes as well, and the variants with the minimal free energies (Table S1) were selected for MD simulations. The complexes and the G4 cores of TBA and F-thio-TBA were stable throughout the simulation. The representative snapshots for (R/S)-F-thio-TBA are shown in Fig. 2B. For other diastereomers, see Supplementary Figs. S7A and S8A. The trajectories are provided as Supplementary Movies.
In the case of the (S)-F-thio-TBA complex with the V3 loop, amino acid residues TYR318 and THR320 played major roles in binding. The GLN330 residue from the V3 loop and the ARG440 residue from the nearby region participated in H-bonding with unmodified TBA, (R)-F-thio-TBA, and (R/S)-F-thio-TBA. In the CD4-binding site, LYS282, LYS487, and ARG480 most significantly contributed to electrostatic interactions and H-bonding with the aptamers. As evident from Fig. 2B and Supplementary Fig. S8A, the disaccharide composed of FUC (alpha-
The estimation of the binding energies by the MM-GBSA method (Supplementary Table S2 and Supplementary Figs. S7B and S8B) revealed that all three analyzed diastereomers of F-thio-TBA were superior to unmodified TBA. Substitution of O for S in the aptamer internucleotide linkages enhanced hydrophobic interactions with the target; the aptamer was drawn into closer proximity and as a result formed more H-bonds with gp120 (Supplementary Figs. S7D and S8D). This result agrees with the data in Tables S1 and S2: the electrostatic binding energy (ΔGelec) is higher in TBA, whereas the van der Waals (ΔGvdW) and total (ΔGtotal) energies are higher in the thio-aptamers.
To summarize, the molecular modeling results suggest that interactions with gp120 may underlie the observed minor anti-HIV activity of TBA (several gp120 binding sites may be involved, including the V3 loop). The thiophosphoryl TBA analog has a higher affinity for the gp120V3 loop. Similar to BclON, TBA and its thio analogs appear to retain the G4 conformation upon binding with gp120.
In conclusion, new potent anti-HIV-1 aptamers were obtained. The G4-forming thiophosphoryl ON F-thio-BclON and the unmodified (presumably non-G4-forming at submicromolar concentrations) ON BclON-mut inhibit lentiviral transduction in the low nanomolar range and thus can be considered for the development of microbicides for HIV-1 prevention. Both ONs most likely act via binding with the HIV-1 gp120 glycoprotein, although other targets cannot be excluded.
Footnotes
Acknowledgments
This work was supported by the Russian Science Foundation (14-25-00013) (physicochemical studies, ON synthesis and molecular modeling), the Dynasty Foundation (physicochemical studies—in part), and the Program of Fundamental Research of RAS “Molecular and Cell Biology” (activity tests).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.