
Abstract
Messenger RNA (mRNA) is a promising new class of therapeutics that has potential for treatment of diseases in fields such as immunology, oncology, vaccines, and inborn errors of metabolism. mRNA therapy has several advantages over DNA-based gene therapy, including the lack of the need for nuclear import and transcription, as well as limited possibility of genomic integration. One drawback of mRNA therapy, especially in cases such as metabolic disorders where repeated dosing will be necessary, is the relatively short in vivo half-life of mRNA (∼6–12 h). We hypothesize that protein engineering designed to improve translation, yielding longer-lasting protein, or modifications that would increase enzymatic activity would be helpful in alleviating this issue. In this study, we present two examples where sequence engineering improved the expression and duration, as well as enzymatic activity of target proteins in vitro. We then confirmed these findings in wild-type mice. This work shows that rational engineering of proteins can lead to improved therapies in vivo.
Introduction
M
Although mRNA therapies have dramatic potential, there are several technical challenges that must be solved before therapies are possible for patients suffering from inborn errors of metabolism or other rare diseases. Nature has evolved strict control over protein production, from transcription and translation regulation, to post-translational modification of the proteins themselves. As part of this process, mRNA is transcribed when needed and degraded shortly thereafter, making mRNA very transient in its nature. Typical mRNA half-life ranges between 6 and 12 h with the median half-life at ∼9 h, and protein produced from this mRNA tends to have half-lives ranging from 1 h to 40 days, with the median half-life at ∼46 h [2]. While there are examples of extreme mRNA and/or protein stability [2] (50 h half-life for mRNA, 1,000 h half-life for proteins), the vast majority of mRNA and proteins are transient–synthesized when they are needed by the cell and degraded shortly thereafter.
To combat the problem of transient mRNA and protein stability, three key areas of optimization should be targeted (Fig. 1). First, safe and efficacious delivery formulations designed to target specific tissues or organs, such as lipid or polymer nanoparticles [3], protein-based polymers [4], or viral-based vectors [5], must be developed with the intention of weekly or biweekly dosing. Patients suffering from rare disorders or inborn errors of metabolism will require dosing for their entire lives. Second, the mRNAs themselves should be optimized to maximize protein translation, mRNA half-life, or both. This may be accomplished by modification of the 5′ cap [6], identification of stabilizing or translation enhancing 5′ or 3′ untranslated regions (UTRs) [7], codon optimization [8], modified nucleotides [9], modified linkages between the nucleotides [10], or the use of preassembling translation initiation complexes in nanoplexes [11]. Care must be taken to ensure efficient translatability, while improving stability and avoiding any sort of toxicity or immune response by the target tissue or organs. Finally, the protein sequences can be engineered to impact protein stability, half-life, and/or enzymatic activity. We believe a combination of these three key areas will result in stable, nontoxic therapies that may be dosed at biweekly intervals, if not at even longer durations.
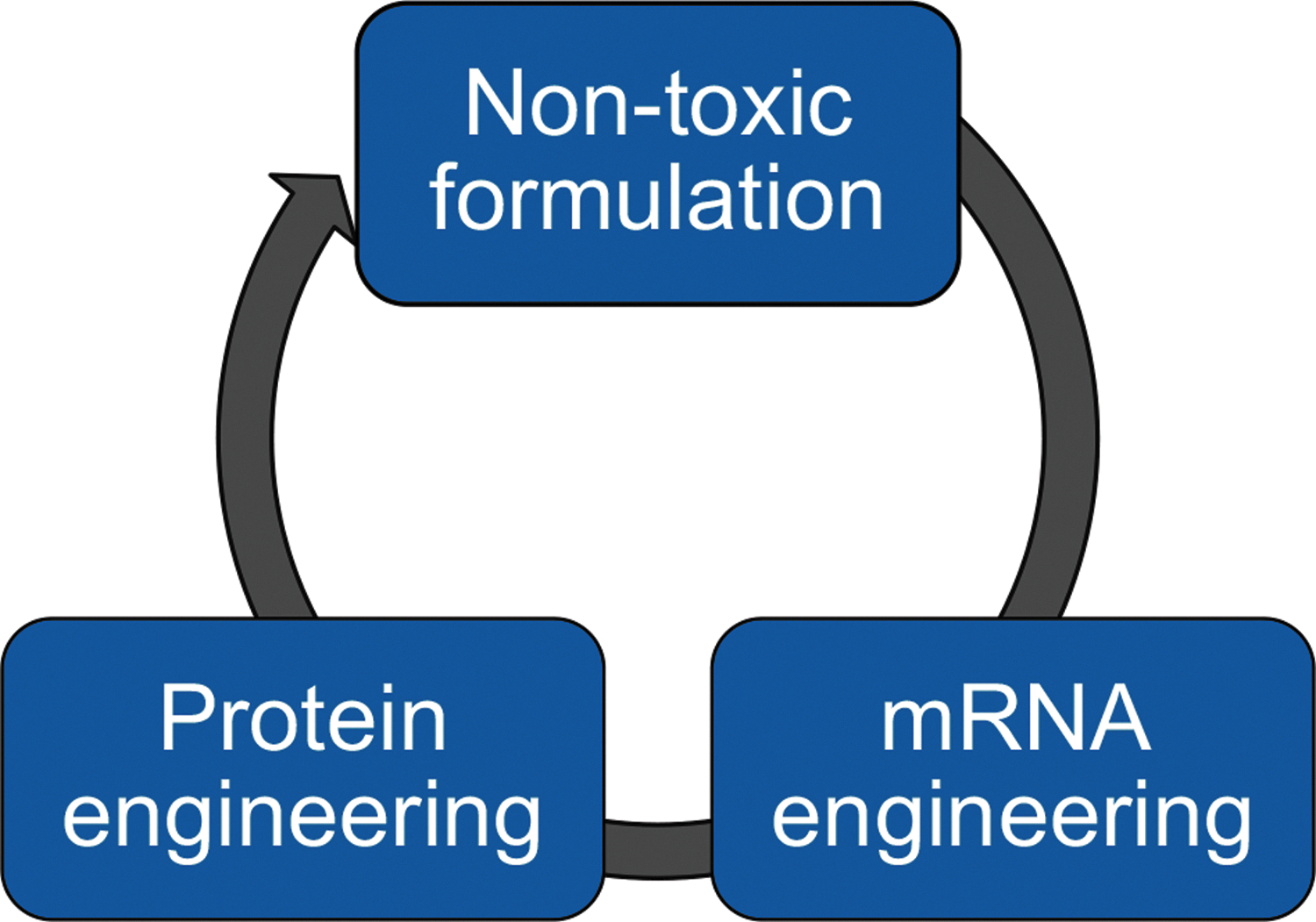
Three key areas of optimization for better mRNA therapies. Due to the transient nature of mRNA and protein, mRNA therapies will need to be optimized to be successful. First, nontoxic formulations must be developed that can target specific tissues of interest safely. Second, the therapeutic mRNAs must be engineered to improve half-life or stability. Finally, the protein expressed from mRNA therapies can be engineered to improve half-life, expression, and/or activity. mRNA, messenger RNA.
In this study, we discuss the third key area—protein engineering. Protein engineering has been used previously to improve protein therapeutics [12–18], but has yet to be utilized in the improvement of protein replacement therapies derived from mRNA therapeutics. We utilize two proprietary protein engineering algorithms designed to improve protein expression or half-life, as well as to improve enzymatic activity. We present two examples where protein engineering improved protein expression and/or activity in cell-based assays and then show improved expression in wild-type mice for one of the examples. We also show correlation between cell-based assays and assays in vivo, which should allow researchers to screen vast numbers of therapeutic candidates before deployment in animal models. This work, in addition to optimized formulations and mRNA engineering, describes methods to optimize and progress mRNA therapies to clinical trials for rare and ultra-rare diseases.
Materials and Methods
Target selection
Two different proteins were chosen for the proof of concept studies by using the following criteria: (i) liver targets with associated well-characterized human disease; (ii) high-resolution crystal structure available; (iii) similar purification scheme for high-throughput protein production; and (iv) easily measurable activity assay using off-the-shelf kits or reagents. The first protein chosen was arginase 1 (ARG1), a trimeric enzyme typically expressed in the cytoplasm of hepatocytes and involved in the final step of the urea cycle [19]. ARG1 converts L-arginine to urea and L-ornithine, and patients deficient in ARG1 present with typical urea cycle disorder symptoms, including hyperammonemia, encephalopathy, respiratory alkalosis, and intellectual disabilities [20]. Patients with arginase deficiency are treated with a combination of nitrogen scavengers and diets that are low in protein. However, patients must be managed carefully for the duration of their lives to ensure balance between intake of proper amounts of protein required for growth, as well as the prevention of hyperammonemic events [21].
The second protein chosen was hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1), a tetrameric enzyme involved in the purine salvage pathway and associated with Lesch–Nyhan Syndrome (LNS). HPRT1 has a broad tissue distribution and catalyzes the conversion of hypoxanthine to inosine monophosphate and guanosine to guanosine monophosphate [22]. Patients suffering from LNS typically have a buildup of uric acid in body fluids. This results in hyperuricemia and hyperuricosuria, as well as poor muscle control and moderate-to-severe intellectual disability. Patients often exhibit self-mutilating behaviors, repetitive movements of the arms and legs, and involuntary writhing [23]. Treatment for LNS is mainly targeted at symptom management and allopurinol can be used to control the levels of uric acid in body fluids [24], but no treatment is effective in controlling the neurological effects of the disease.
Protein engineering
Two engineering protocols optimized in-house at Alexion Pharmaceuticals, Inc. were employed to design variants of each targeted protein. The algorithms were designed to improve protein thermostability, expression, and/or to improve the enzymatic activity in comparison to wild-type protein. The first protocol involved use of the consensus-based approach for protein engineering coupled with computational variant selection. Briefly, the human sequences of ARG1 and HPRT1 were downloaded from UniProt. Basic Local Alignment Search Tool was used to identify sequences of closely related orthologs. Sequences were then aligned using CLUSTALW [25]. Next, consensus frequency at each position was calculated as previously described [26,27]. The residues that were identified were subjected to computational energetics calculations using the Calculate Mutation Energy (Stability) module in Discovery Studio 2017 [28] and the reported X-ray crystal structures of both proteins (ARG1–PDB ID: 2PHA; HPRT1–PDB ID 1Z7G) [19,22]: where residues at each identified position were mutated to each possible residue contained in the consensus sequence. The combination representing the highest calculated stability (most negative ΔΔG) was selected for biochemical and biophysical studies. To optimize the enzymatic activity, surface lysine residues were modified to arginine. This idea was based on previous work that described the acetylation of lysine residues to depress the enzymatic activity. We hypothesized that replacement of lysine with arginine would prevent acetylation and would maintain the enzymatic activity over a longer period [29]. The variants from both approaches differed only in amino acid sequence. Nucleotide sequence, or codon optimization, was not examined in this study. In addition, 5′ or 3′ UTR sequences were not modified. Constructs labeled Variant 1 were designed for improved thermostability, while constructs labeled Variant 2 were designed for greater enzymatic activity.
Design and synthesis of DNA vectors
For bacterial expression of recombinant protein, plasmids expressing open reading frames (ORF) of proteins of interest or their variants were synthesized by Genewiz and subcloned into a modified pET-15b vector using the NdeI and XhoI sites. This particular pET-15b vector replaces the thrombin site with a TEV cleavage site for removal of the N-terminal 6xHis-tag. Clones were also prepared of the ORFs with uncleavable C-terminal myc-FLAG tags for the purpose of standardization for the homogeneous time-resolved fluorescence (HTRF) assay. The ORF sequences were based on the native human nucleotide sequences and were not codon optimized for expression in Escherichia coli. For in vitro transcription of mRNA, plasmids containing a T7 promoter, a 5′ UTR, the ORFs from above, a C-terminal myc-FLAG tag, a 3′ UTR, a synthetic polyA tail of ∼100 nucleotides, and a SapI linearization site were synthesized and scaled up by ATUM. The sequences of the vector, 5′ and 3′ UTRs, polyA tail, and SapI linearization site were exactly as described previously [30].
Expression and purification of target proteins
For expression, chemically competent T7 Express (New England Biolabs) E. coli were transformed with 100 ng of plasmid encoding for each protein or its variant and 100 ng of pGRO7, a plasmid harboring the ORFs for the GroEL/ES chaperones under the control of the araB promoter (Clontech). Transformed bacteria were grown overnight on Luria Broth (LB) agar media with the appropriate antibiotics for selection (carbenicillin at 100 μg/mL and chloramphenicol at 35 μg/mL). The following day, single colonies were inoculated into 10 mL starter LB cultures containing carbenicillin and chloramphenicol and grown overnight. The following morning, 1 mL of overnight culture was inoculated into a 1 L LB culture containing carbenicillin, chloramphenicol, and 0.5 mg/mL L-arabinose (for induction of GroEL/ES). Samples were grown to an OD600 of 0.8 at 37°C, at which point, the temperature was dropped to 28°C and 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to induce the expression of the target proteins. The cultures were grown for 16 h at 28°C. Cells were pelleted and frozen at −80°C until processing. To purify the proteins, bacterial pellets were thawed on ice and 5 mL of lysis buffer per gram of pellet was added. The lysis buffer consisted of 1× phosphate-buffered saline with 1 mg/mL of lysozyme, 1× BugBuster (EMD Millipore), 0.0625 mg/mL DNaseI (Sigma), and 1 tablet of SigmaFast Protease Inhibitor (Sigma). Pellets were resuspended manually by pipette and then rotated at 4°C for 30 min before the pelleting of cellular debris at 100,000 g. Protein was purified from the supernatant using tandem affinity column chromatography on an ÄKTA Pure FPLC (GE Lifesciences). The first column was a HisTrap column (GE Lifesciences) equilibrated with PBS and an elution buffer consisting of PBS plus 1 M imidazole. The second column was a HiTrap Q anion exchange column (GE Lifesciences) equilibrated with 25 mM Tris pH 7.5 and an elution buffer consisting of 25 mM Tris pH 7.5 plus 1 M NaCl. Fractions from the HisTrap column were pooled and diluted to 50 mL in 25 mM Tris pH 7.5 before loading onto the HiTrap Q column. Fractions from the HiTrap Q column were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to ensure >99% purity and pooled. The 6xHis-tag from each protein was removed with TEV protease at room temperature overnight in a buffer consisting of 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT, and 5% glycerol. The His-tagged TEV protease was removed from the samples by running the mixture over a HisTrap column and collecting the flow-through. Flow-through fractions were analyzed by SDS-PAGE to ensure purity. Suitable fractions were combined and concentrated. Protein samples were stored at −80°C in small aliquots until use.
Thermal shift assays
Protein thermal shift assays were performed using a StepOnePlus real-time polymerase chain reaction system equipped with StepOne software v2.3 and Protein Thermal Shift software v1.2 (Applied Biosystems). Protein samples were removed from −80°C storage, thawed on ice, and pelleted to remove any aggregated protein. Protein concentration was determined in triplicate by NanoDrop 8000 (Thermo Scientific) using the expected molecular weight and extinction coefficient. Proteins were diluted to a concentration of 20 μM in PBS supplemented with Sypro Orange (Life Technologies; 1:5,000). Twenty μL of each protein sample was aliquoted in replicates of 12 into MicroAmp Fast Optical 96-well plates and sealed with MicroAmp Optical Adhesive film (Thermo Scientific). Samples were incubated at 25°C for 5 min before initiation of the heat ramp step (set to 1%, which is approximately 1.3°C/min) from 25°C to 95°C with constant fluorescent monitoring. Melting curves and Tms were calculated using the StepOne and Protein Thermal Shift software packages.
Activity assays
For ARG1 assays, the arginase activity assay kit (Sigma) was used according to the manufacturer's recommendation. For HPRT1 assays, the PRECICE HPRT assay kit (Novocib) was used according to the manufacturer's recommendation.
In vitro transcription of mRNA
mRNA was produced as described previously [30,31]. Briefly, T7 RNA polymerase was used to transcribe mRNA from SapI-linearized plasmid DNA template encoding a 5′ and 3′ UTR, as well as polyA tail of ∼100 nucleotides. More than 85% 7mGppp-capping (Cap 0) was generated through incorporation of cap analog and lowered GTP concentration in the transcription reaction. Modified UTP (1-methylpseudouridine triphosphate) replaced UTP in the transcription reaction at 100%. Transcription reactions were run for 3 h at 37°C and immediately diluted into a DNase I reaction at 37°C for 1 h. Transcripts were purified by oligo-dT affinity purification, and the 5′-cap was methylated with 2′-O-methyltransferase to generate Cap1 mRNA. Finally, the mRNA was purified over a C18 RP-HPLC column.
Cell Culture
The human cervical carcinoma HeLa cell line was obtained from American Type Culture Collection (ATCC). The cells were cultured in an Eagle's minimum essential medium (GIBCO) supplemented with 10% fetal bovine serum and maintained at 37°C with 5% CO2 in a humidified atmosphere. Primary human hepatocytes (PHH) were obtained from In Vitro Admet Laboratories (iVAL). Cells were cultured in a hepatocyte induction medium (HIM) supplemented with the HIM supplement kit (iVAL).
Transfection
HeLa cells were seeded in 24-well tissue culture-treated plates. Twenty-four hours later at >90% confluency, HeLa cells were transfected with mRNA. Each plate contained three wells that were not transfected (cells only), three wells that were transfected with an mRNA encoding enhanced green fluorescent protein (eGFP) (GFP control), as well as three wells transfected with the wild-type proteins and their variants. The amount of mRNA was determined for each target through a titration optimization to determine the half-maximal expression of protein for each cell type examined (Supplementary Figs. S1 and S2; Supplementary Data are available online at www.liebertpub.com/nat). mRNA was diluted in optimal minimal essential medium (OPTI-MEM) serum-free media (GIBCO) (30 ng per well for ARG1 construct and 80 ng per well for HPRT1 constructs) and was added to 2 μL Lipofectamine 2000 (Life Technologies) (which was diluted in OPTI-MEM). The mRNA-lipid complexes were allowed to incubate for 15 min at room temperature before adding to cells. Cells were returned to a humidified incubator set to 37°C with 5% CO2. iVAL PHH were ordered preseeded in 24-well tissue culture-treated plates. Media were changed and cells were allowed to recover for 2 h before transfection. PHH were transfected with mRNA. The amount of mRNA was determined for each target through a titration optimization. mRNA was diluted in OPTI-MEM serum-free media (GIBCO) (50 ng per well for ARG1 constructs and 200 ng per well for HPRT1 constructs) and was added to 3 μL mRNA-In (MTI-GlobalStem) (which was diluted in OPTI-MEM). The mRNA-lipid complexes were allowed to incubate for 20 min at room temperature before adding to cells. Cells were returned to the humidified incubator set to 37°C with 5% CO2.
Cell lysis and protein quantification
Cells from both experiments were harvested 24, 48, and 72 h posttransfection to examine the overall expression for each protein and its variants. At appropriate time points, cells were removed from incubation, media were removed, and the cells were washed twice with ice-cold PBS. One hundred microliter of a buffer consisting of 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 1 × Halt Protease and Phosphatase inhibitor cocktail (Thermo Scientific) was then added to each well. The plates were sealed and stored at −80°C until processing. At processing, the plates were removed from −80°C, thawed at 4°C for 30 min, refrozen for 30 min at −80°C, and then thawed once more at 4°C for 30 min. Cell lysates were then mixed manually by pipette before filtration through a 96-well AcroPrep 3 μm GF/1.2 μm Supor Filter Plate (PallProtein concentration in the filtrate was determined in quadruplicate by BCA Assay (Thermo Scientific) in a 384-well format. Briefly, 4 μL of filtrate was added to 100 μL of BCA reagent (49:1 ratio of component A: component B) and incubated for 30 min at 37°C. Next, the absorbance was determined at 560 nm and a standard curve consisting of BSA (from 15.6 to 2,000 μg/mL) was used to determine the protein concentration for each sample.
Target protein quantification from in vitro transfections
To quantitate proteins expressed in HeLa cells, an HTRF was used (Cisbio Bioassays). HTRF assays combine fluorescence resonance energy transfer (FRET) technology with time-resolved measurement to dramatically increase specific signal, while minimizing buffer, media, and cell-based fluorescence interference [32]. The specific HTRF kit used was the FLAG tag kit, which is a competition-based assay where an anti-FLAG antibody labeled with a Eu3+-cryptate donor triggers a FRET transfer to a FLAG-tagged peptide labeled with a d2 acceptor, while in close proximity. FLAG-tagged protein present in samples competes with the interaction between the two conjugates and prevents FRET in a quantitative manner. The FLAG-tag kit was calibrated using recombinant ARG1-FLAG and HPRT1-FLAG giving a detection range in solution of 100 pM–10 nM for ARG1 and 10 pM–1 nM for HPRT1 (Supplementary Fig. S3). For analysis of in vitro transfected cells, lysate concentrations were normalized to 50 μg/mL. Ten microliter of normalized lysate was added to 10 μL of a 1:1 mixture of Eu3+-cryptate anti-FLAG antibody to FLAG-tagged peptide labeled with d2. Mixtures were incubated and read according to manufacturer's recommendation.
Immunofluorescence and imaging
At set time points, cells were washed thrice with PBS and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min at room temperature. Cells were washed thrice with PBS and permeabilized with 0.5% Triton-X100 (Sigma) for 10 min. Cells were washed thrice with PBS and then blocked in 10% normal horse serum (NHS) (Vector Laboratories) for 1 h at room temperature. Primary antibody, FLAG-tag Rabbit mAb (Cell Signaling Technologies), was used at 1:100 diluted in 2% NHS and incubated overnight at 4°C on a shaker platform. The secondary antibody used was an Alexa Fluor488 goat anti-mouse (Molecular Probes) at 1:1,000 diluted in 2% NHS and incubated for 1 h at room temperature on a shaker platform and protected from light. For cell identification, nuclei were stained using NucBlue Fixed Cell ReadyProbes (Molecular Probes). Two channel images were acquired simultaneously using the 20× air objective on the Perkin Elmer Operetta Confocal, LED, sCMOS (CLS) system. Six fields of view were imaged per well. To quantify FLAG-tagged protein expression, Harmony High Content Imaging and Analysis software was used to segment nuclei and cytoplasm and calculate mean fluorescent intensity of the green channel per cell.
Animal studies
Animals were housed and handled according to institutional guidelines, and experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) review board. C57BL/6 animals (8-week-old male mice) were obtained from Jackson Laboratories (Bar Harbor, ME). Mice were kept in a temperature-controlled environment with a 12-h light /12-h dark cycle, with a standard diet and water ad libitum.
Formulation of mRNA into lipid nanoparticles and in vivo dosing
mRNA constructs were formulated using a self-assembling lipid nanoparticle (LNP) containing either wild-type human HPRT1 or its engineered variants. A GFP encoding mRNA was used as an mRNA-loaded vehicle control and PBS was used as a negative control in this experiment. LNPs were prepared by rapid mixing of mRNA in acetate buffer at pH 4.0 with a proprietary lipid mix from Precision NanoSystems, suspended in ethanol at a lipid-to-drug ratio of ∼11 (wt/wt). LNPs were immediately dialyzed and 0.2 μm sterile filtered. A table with characterization properties of the LNPs can be found in Supplementary Table S1. Animals were injected with a single intravenous dose of 1 mg/kg of formulated mRNA through the tail vein. Blood samples were collected by submandibular bleed pretreatment and by terminal cardiac puncture at 24, 48, 96, and 168 h postinjection for analysis of liver transaminases (n = 5 per time point). Serum samples were stored at −80°C till analysis. Levels of liver transaminases alkaline phosphatase (ALP), alanine transaminase (ALT), and aspartate aminotransferase (AST) (Beckman Coulter) in serum were measured using AU480 clinical chemistry analyzer (Beckman Coulter). Liver samples were harvested for assaying hepatic expression, frozen in 7 mL Precellys tubes, and stored at −80°C until processing (n = 5 per time point).
Liver tissue homogenization, lysate concentration determination, and target protein quantification
Whole livers (∼1 g) were removed from storage and thawed on ice for 20 min. Four milliliter of lysis buffer consisting of 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 1 × Halt protease and phosphatase inhibitors (Thermo Scientific) was added to each liver sample. Samples were homogenized using 5, 20 s pulses with 20 s pauses at 6,800 revolutions per minute using a Precellys Evolution tissue homogenizer. Homogenate was transferred to 5 mL centrifuge tubes (Eppendorf) and centrifuged at 21,191g for 30 min at 4°C in an FA-45-16-17 rotor using an Eppendorf 5430R tabletop centrifuge. Supernatants were carefully transferred to new tubes and stored on ice. Lysate protein concentration was determined in quadruplicate by the BCA Assay (Thermo Scientific) in a 384-well format as previously described. Target protein quantification was performed by anti-FLAG HTRF assay as described previously with liver lysates normalized to 20 mg/mL. Leftover liver lysates were stored in aliquots at −80°C.
Data analysis and statistics
Relevant data and statistical analysis were carried out using Microsoft Excel and GraphPad Prism. Group comparisons were made using a standard two-way ANOVA followed by Tukey's post-hoc analysis versus reference (reference was generally wild-type protein). Statistical significance was set at P ≤ 0.05. All charts or graphs displayed in this article were constructed using GraphPad Prism.
Results
Protein engineering
The main aim of these studies was to determine whether modification of a protein's sequence could improve expression, stability, and/or activity in cell culture, and eventually in vivo. Two underlying hypotheses were examined. First, we theorized that amino acid modifications designed to increase the thermal stability of the proteins would lead to increased cellular expression. Second, we theorized that modifications to surface residues of the proteins could increase the relative enzymatic activity of each target.
To increase protein stability, we employed the consensus-based approach described previously [27] with the addition of computational-based selection of the final sequence. A detailed description of methods used is in the Materials and Methods section. The top ten sequences with the most negative ΔΔG was selected for inspection before selection of the top candidate for biophysical and biochemical characterization. For ARG1, consensus frequency analysis suggested residues A3, R6, T7, V30, I58, S77, K83, G119, R205, and L306 for analysis (Fig. 2A). Computational- and human-based analysis narrowed the final modifications to V30Y and K83V (Fig. 2C). For HPRT1, the same analysis suggested residues G7, N26, R34, F36, V53, Q144, V150, R151, K175, and V205 with final modifications of N26K, R34K, V53I, R151K, and V205I for further testing (Fig. 2B, D).
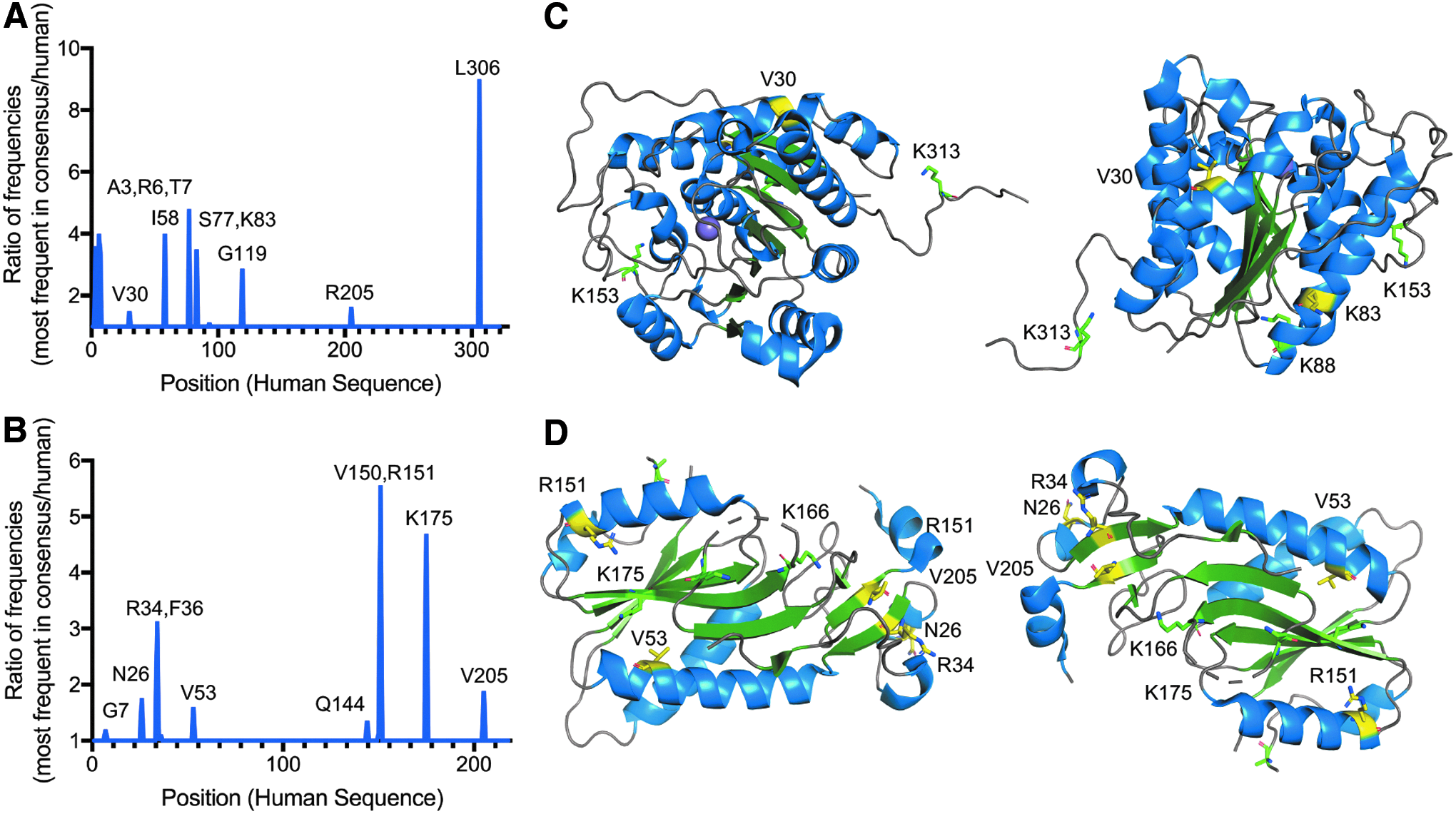
Engineering of ARG1 and HPRT1.
Many metabolic enzymes are posttranslationally modified (PTM) to control their activity. In one type of PTM, surface lysines are acetylated to decrease the enzymatic activity [29]. In one study, modification of a surface lysine of argininosuccinate lyase to arginine resulted in an enzyme that was 132% as active as wild type [29]. To test whether this approach would work for ARG1 and HPRT1, surface lysines were selected and modified to arginine. For ARG1, K4, K88, K153, K313, and K322 were selected for modification and for HPRT1, K115, K159, and K166 were chosen (Fig. 2C, D).
Recombinant production of ARG1, HPRT1, and their variants
To test our engineering approaches, we first designed and synthesized bacterial expression vectors harboring genes for our two target proteins, ARG1 and HPRT1, as well as their engineered variants for thermostability and activity. The proteins and their variants were expressed and purified to >99% purity (with the exception of a small, presumed degradation product for HPRT1 variant 1, which did not interfere with downstream biophysical or biochemical assays) (Fig. 3A, B).
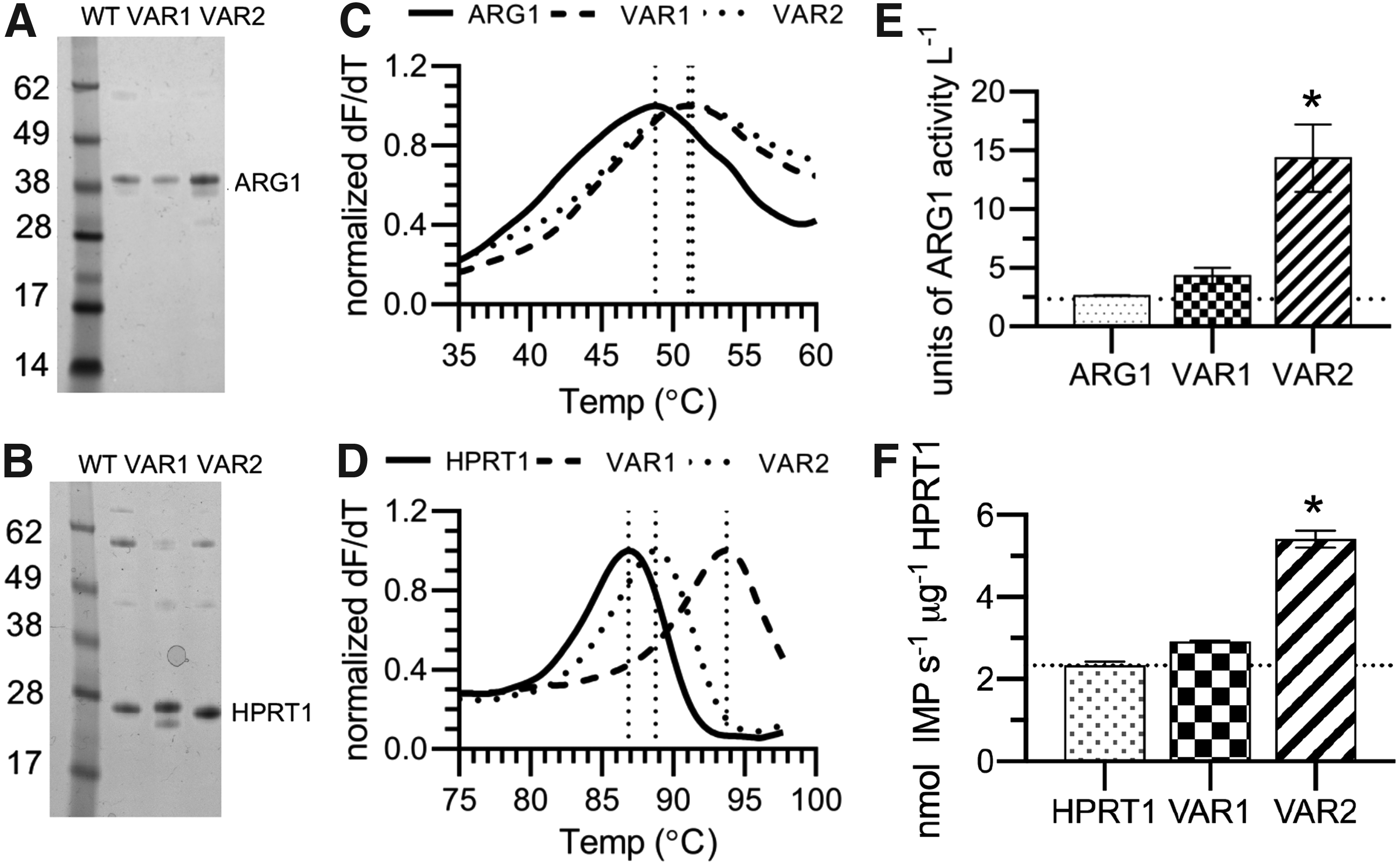
Characterization of recombinant ARG1, HPRT1, and their engineered variants.
Thermal stability and enzymatic activity of the proteins and their variants
To examine the effect of the amino acid modifications on thermostability, differential scanning fluorimetry [33,34] was performed on each protein construct. The melting temperature (Tm) was calculated for each construct by plotting the derivative ratio between fluorescence and temperature (dF/dT). All constructs examined underwent simple, two-state transitions where a single Tm for each construct could be calculated [33]. For wild-type ARG1, the Tm was determined to be 48.7°C. For the ARG1 variant designed to be more thermostable, a 2.4 degree upward shift in Tm to 51.1°C was noted. In addition, a 2.7 degree upward shift in Tm to 51.4°C was noted in the variant designed to have higher enzymatic activity (Fig. 3C). For wild-type HPRT1, the Tm was determined to be 86.8°C. For the HPRT1 variant designed to be more thermostable, a 6.9 degree upward shift in Tm to 93.7°C was seen, while in the variant designed to be more enzymatically active, a 2 degree upward shift in Tm to 88.8°C was noted (Fig. 3D). The increase in stability for both variants designed to have higher enzymatic activity was unexpected. However, previous studies have noted stability and stability-function tradeoffs in protein adaptation by conservative mutations [35]. It is possible that the modification of lysine residues to arginine (and the hypothesized decrease in acetylation) could contribute to the higher stability in these variants. Further work would be necessary to elucidate the nature of the stabilizing effects noted in this study.
To examine the possible effect of the amino acid modifications on enzyme activity, the specific activity was calculated for each protein and its variants using a relevant assay with the recombinant protein prepared previously. For ARG1 and its variants, an arginase activity assay kit (Sigma) was used. Wild-type ARG1 and the variant designed to be more thermodynamically stable were determined to have similar levels of activity. We found a significant increase in activity for the variant designed to have higher activity compared to wild type (5.5-fold increase) (Fig. 3E). For HPRT1 and its variants, the PRECICE HPRT assay kit (NovoCib) was used. The variant designed to be more thermodynamically stable had a 1.3-fold increase in activity over wild-type HPRT1. In addition, the variant designed to have a higher activity had a 2.3-fold increase in activity over wild-type HPRT1 (Fig. 3F). These initial experiments with recombinant proteins confirm that protein engineering can be utilized to improve thermal stability and increase enzymatic activity in vitro. We next asked whether similar trends would be seen in cell-based assays.
Cell-based expression and activity assays of the proteins and their variants
To examine the effect of protein engineering on expression and activity in a cell-based system, mRNAs encoding the proteins and their variants with C-terminal FLAG tags were transfected in HeLa cells. HeLa cells were chosen as an initial screening platform for several reasons: (i) their ease of growth in cell culture, (ii) their ability to grow for extended periods of time, and (iii) their lack of expression of the two target proteins chosen, allowing for an accurate analysis of non-endogenous expression and activity. Proteins contained C-terminal FLAG tags for ease of detection and quantification using an anti-FLAG HTRF assay.
The amount of FLAG-tagged protein per gram of HeLa lysate was calculated for each wild-type construct and engineered variant at each time point using calibrated standards. Expression of each protein was maximum 24 h posttransfection. For ARG1 and its variants at 24 h, the variant designed to have higher thermostability had an approximately threefold increase in expression over wild type, while the variant designed to have higher activity expressed at only 26% the level of wild type. This trend generally continued over the next two time points, with the thermostable variant expressed at 2.8- and 2.4-fold over wild type at 48 and 72 h, and the variant designed to have higher activity expressed at 25% and 30% that of wild type (Fig. 4A). For HPRT1 and its variants, the variant designed to have greater thermostability had 11.8-, 11.6-, and 13.8-fold increases in expression over wild type at 24, 48, and 72 h. The variant designed to have greater activity also had higher expression than wild type, 1.4-, 1.4-, and 1.2-fold increase over wild type at 24, 48, and 72 h, respectively (Fig. 4B). No expression was noted for GFP-transfected cells or wells where no transfection reagents were added.
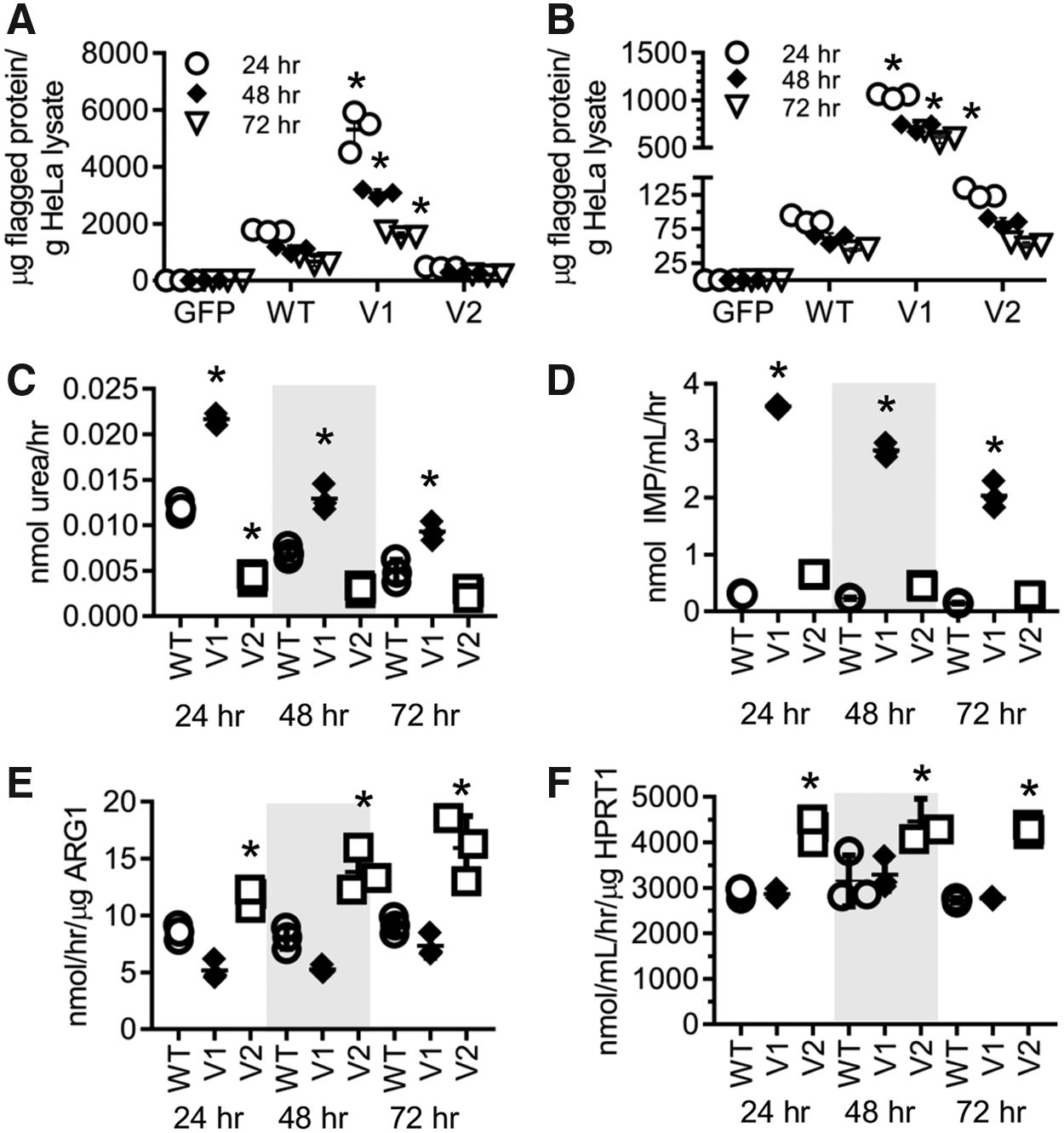
Cell-based expression and activity assays in HeLa cells.
Next, we examined enzymatic activity for the constructs using the activity assays used for determining activity of the recombinant proteins. A general trend was noted for activity, where lysates having greater levels of protein expressed had greater enzymatic activity (Fig. 4C, D). To determine whether the variants had an effect on the amount of activity, overall activity levels were normalized to protein expression. For ARG1, cells expressing variant 1 had on average 68% the level of activity of wild type, and cells expressing variant 2 had on average a 1.6-fold increase in activity (Fig. 4E). For HPRT1, cells expressing variant 1 had on average an equivalent level of activity compared to wild type, and cells expressing variant 2 had on average a 1.5-fold increase in activity (Fig. 4F).
To confirm the results we saw with the HTRF assays, and that the proteins and their variants were correctly localized to the cytoplasm, we examined the expression of the constructs in HeLa by microscopy using the Perkin Elmer Operetta CLS High-Content Analysis System. HeLa cells were seeded on glass-bottom 24-well plates and transfected as described for the expression assays. Diffused AF-488 (green) signal was observed in the cytoplasm of cells (Fig. 5A, B). The eGFP-transfected cells showed complete cell transfection at the concentration tested (data not shown), suggesting that treated cells with lower fluorescence intensity was due to the protein construct expression level and not transfection. For ARG1 and its variants, wild type and variant 1 showed comparable levels of expression, and both expressed higher levels than variant 2 (Fig. 5C). This was contradictory to the previous assays where variant 1 had a 3-fold increase in expression over wild type. Perhaps this represents inaccessibility of the FLAG tag in the ARG1 variant 1 construct, or perhaps suboptimal staining conditions were present. For HPRT1 and its variants, variant 1 showed the highest level of expression (Fig. 5D), which concurred with the previous studies.
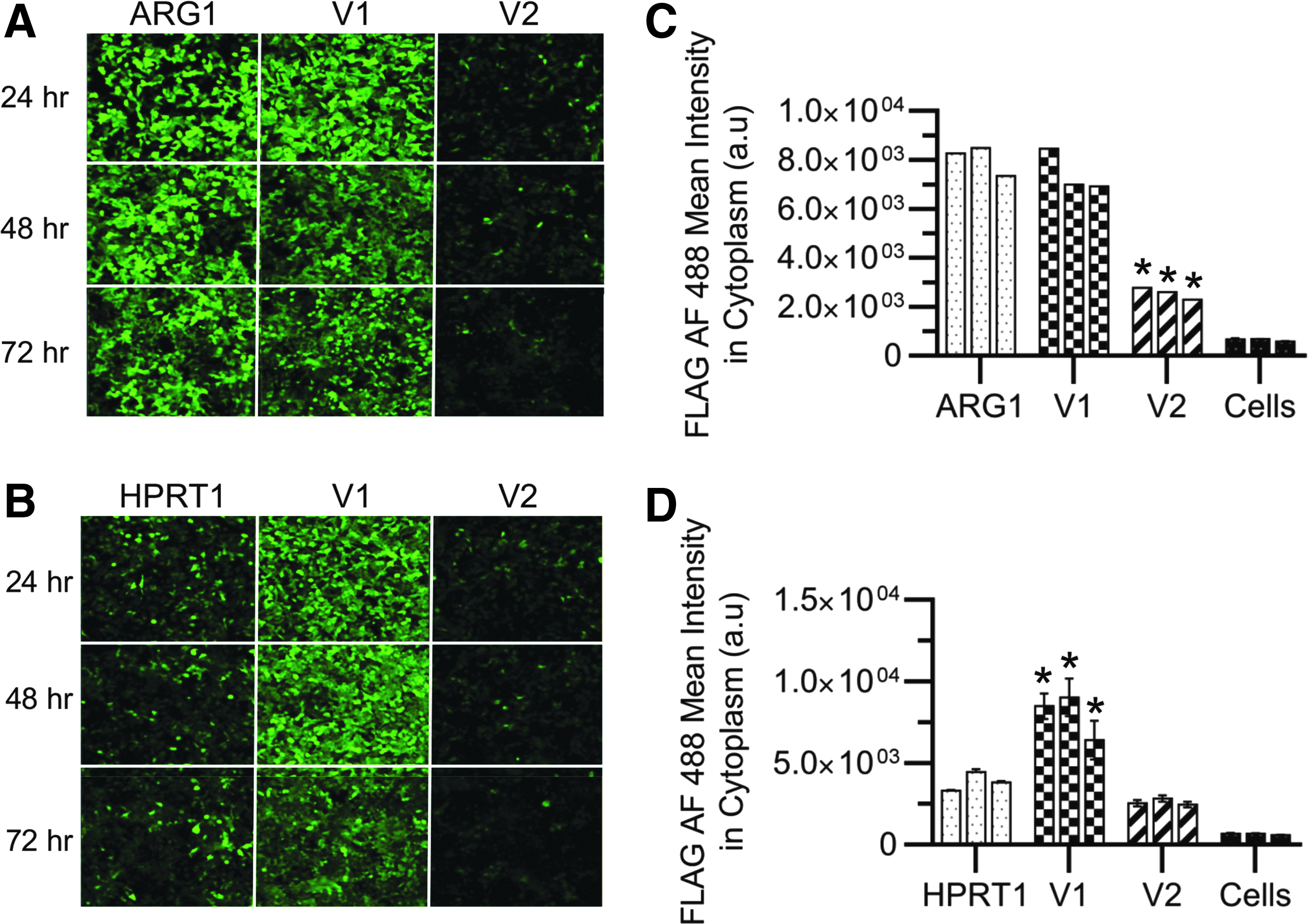
Immunofluorescence analysis of ARG1, HPRT1, and their variants. Immunofluorescence staining of different FLAG-tagged protein variants of
To examine whether the expression trends seen in HeLa would reproduce in a relevant cell line, we next examined the expression of the proteins and their variants in PHH. The PHH were purchased from iVAL preplated in 24-well plates. As with the HeLa cells, expression of each protein was maximum 24 h posttransfection. For ARG1 and its variants, the variant designed to have higher thermostability had an approximately twofold increase in expression over wild type, while the variant designed to have higher activity expressed at only 28% the level of wild type. (Fig. 6A). For HPRT1 and its variants, the variant designed to have greater thermostability had an approximately fivefold increases in expression over wild type at all time points. The variant designed to have greater activity also had higher expression than wild type, an ∼1.4-fold increase over wild type at 24, 48, and 72 h (Fig. 6B). No expression was noted for GFP-transfected cells or wells where no transfection reagents were added. These results are largely consistent with what was seen in HeLa cells. Activity analysis was not performed given the high levels of endogenous ARG1 and HPRT1 in hepatocytes. Future studies could make use of the FLAG tag to pull down the expressed protein and assess activity.
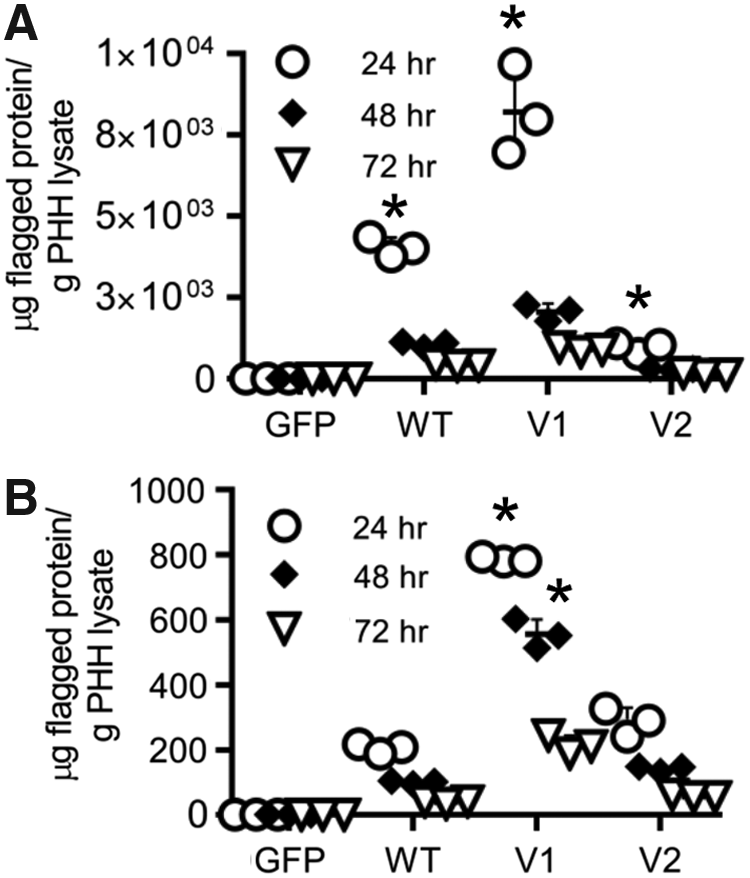
Cell-based expression and activity assays in primary human hepatocytes.
Expression assays of HPRT1 and its variants in wild-type mice
As expected, [26,27] these results indicate that protein engineering can have an effect on either expression or activity in cell-based assays. The variants designed to have higher thermostability expressed more protein than their wild-type counterparts for both ARG1 and HPRT1. The variants designed to have higher activity also have a higher specific activity than their wild-type counterparts for both proteins, but gave varying results in their expression. ARG1 variant 2 gave lower expression than wild type, while HPRT1 variant 2 expressed slightly better than wild type. These results were surprising, but upon consideration, it is possible that the surface modifications introduced to ARG1 may have increased overall degradation. The substitutions may have exposed a protease site, or led to the activation of a degradation pathway. Further studies would be required to understand the specific cause of the increased degradation in ARG1 variant 2. Based on these results, both types of engineering give some degree of flexibility when designing mRNA-based therapeutics. In diseases where a large amount of protein will be necessary, thermostability-based engineering would be beneficial, whereas in diseases where a high degree of activity will be necessary, activity-based engineering would provide benefit. To date, no studies have shown that engineered protein delivered by LNP-formulated mRNA provides a longer duration or higher expression of protein. To determine whether the increases in expression seen in cell-based assays would translate in vivo, we examined the expression of these constructs in wild-type mice. We chose to examine HPRT1 and its variants due to the larger increases in expression seen for variants of both types of engineering approaches. Unfortunately, enzyme assays could not be performed given the high level of endogenous HPRT1 in wild-type animals. True analysis of the activity of these variants will require an hprt1-deficient mouse model or selective pull-down of the expressed protein by the encoded FLAG tag.
We delivered GFP mRNA, HPRT1 mRNA, or its variant mRNAs formulated in ionizable cationic LNPs through a single-dose i.v. injection in male, (8-week old) C57BL/6 mice. LNPs encapsulating different HPRT1 constructs were produced with similar physicochemical characteristics: diameter of ∼88–95 nm, polydispersity index of 0.12–0.14, and encapsulation efficiency of ∼98% (Supplementary Table S1). Serum liver transaminases were measured pretreatment and at 24, 48, 96, and 168 h after dosing to assess any hepatotoxicity observed is specific to the variants when compared to wild-type HPRT1. ALP levels were within normal range for wild-type mice through the time course. Some elevations in ALT and AST levels were observed, but they were not specific to HPRT1 and its variant groups (some elevations seen in GFP vehicle control group), and could be attributed to the formulation and/or high treatment dose concentration (1 mg/kg) (Supplementary Fig. S4). Liver expression of the proteins was monitored by HTRF assay at four time points after dosing (24, 48, 96, and 168 h). Protein expression is detectable at 24 h, with wild-type HPRT1 and variant 2 being roughly equivalent, while variant 1 is expressed at roughly 2.1-fold the level of wild type (Fig. 7A, B). Expression of wild-type HPRT1 and variant 1 peaks at 48 h (Fig. 7C) with the engineered variant expressing ∼2.6-fold more protein than wild type. By 96 h, HPRT1 and variant 1 are roughly equivalent (Fig. 7D). Expression of variant 2 peaks at 24 h and decays from there, and by 48 h, the expression is ∼10% of wild type (Fig. 7B–D). No expression is detectable for any variant by 168 h.
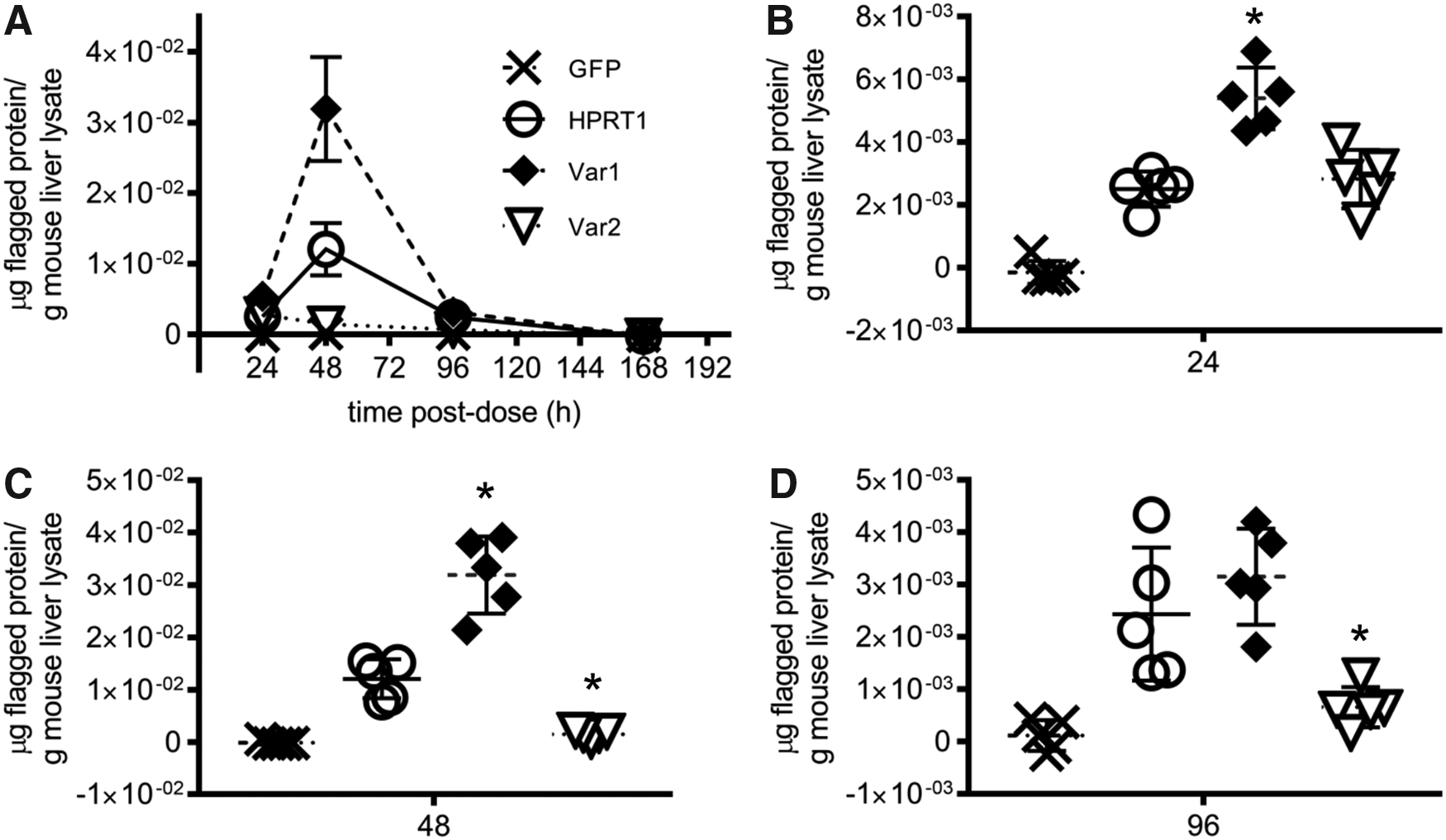
Lipid nanoparticle-formulated HPRT1 or HPRT1-variant mRNA expresses protein in mouse liver. Data are presented as the mean and standard deviation of expression level (n = 5 animals per group).
Discussion
mRNA-based therapeutics are an attractive modality for a wide range of diseases because of their enormous flexibility over traditional biologic and enzyme replacement therapeutics. mRNA-based therapeutics utilize the host cell's endogenous machinery for the production and posttranslational modification of target proteins, allowing the development of therapeutics for diseases not previously targetable. Although mRNA-based therapeutics have immense potential to treat a wide range of diseases, they are not without issue. mRNAs typically have very short half-lives (6–12 h), and the protein produced have half-lives ranging from 1 h to 40 days [2]. In fact, in one report where LNP-formulated mRNA encoding erythropoietin was systemically delivered to rhesus macaques, expression of the target protein was undetectable within 72 h of dosing [36]. Dosing patients at this frequency requires consideration of patient safety and compliance. To combat the problem of transient mRNA and protein stability, we believe three key areas of optimization should be targeted (Fig. 1). First, well-tolerated and nontoxic delivery formulations must be designed and implemented. In this study, we used LNPs, a technology that has been used to deliver siRNA in the clinic and is emerging as a delivery modality for mRNA in the clinic [37,38]. Second, the mRNA should be modified to improve either stability or the translation of protein. Modifications could include sampling of new types of 5′ caps, identification of stabilizing or translation enhancing 5′ or 3′ UTRs, codon optimization to improve translation, modified nucleotides, modified nucleotide linkages, or the use of formulated, preassembled translation initiation complexes. In this study, we discuss the third area—protein engineering. We utilize two protein engineering protocols designed to improve protein stability (enhancing expression and/or half-life) or enzymatic activity.
In this study, we have confirmed that protein engineering can accomplish either increased expression or increased enzymatic activity in two separate cases. We evaluated protein engineering of two proteins, ARG1 (the terminal enzyme in the urea cycle, which is deficient in patients with argininemia [20]) and HPRT1 (involved in the purine salvage pathway and associated with LNS [24]). To date, protein engineering has been utilized to produce therapeutics to treat a wide variety of indications, including cancer, inflammation, autoimmunity, and genetic disorders [12]. These therapeutics range from humanized antibodies [17], enzymes for metabolic disorders [15,16], antibody-drug conjugates [13], and proteins that involve glycoengineering [14,18]. The engineering in these cases typically improve protein production, product safety or efficacy, or improvement in pharmacodynamics or pharmacokinetics [12]. In this study, we examine whether protein engineering can improve the outcome of mRNA-derived therapies and describe methods to engineer and screen sequences in cell-based assays and animals. We first designed sequences and then produced recombinant protein for biophysical and biochemical assays. Upon confirming the desired outcome (either increased thermostability or increased enzyme activity), we checked for increased expression or activity in cell-based assays. For both enzymes examined, the variants designed for thermostability expressed to a higher degree than the wild-type protein. As for the variants designed for higher enzymatic activity, the ARG1 variant expressed at lower levels than wild-type and the HPRT1 variants expressed slightly above the level of wild type. The fact that proteins designed to be more thermostable express at higher levels is not surprising. However, the reason for the higher expression is unclear. Initially, we hypothesized that a more thermostable protein would be more stable in mesophilic cell culture and would resist degradation due to the stability. Another possibility is that thermostability improves protein folding at translation, thereby yielding more protein per unit mRNA introduced. One can conceive a hypothetical situation where perhaps 90% of the protein translated from an mRNA is correctly folded after translation. Perhaps introducing modifications to the protein sequence to improve thermostability may improve this rate to nearly 100%. This may account for the increased protein produced from the sequences designed to be thermodynamically stable. However, this is only a hypothesis and the exact biophysical properties of expression and degradation remain to be studied.
Sequences designed for increased enzymatic activity had much more variable results. In cell-based assays, ARG1 variant 2 expressed to a lower level than wild type, in both HeLa cells and PHH (Figs. 4A, 6A). The opposite was true for HPRT1 variant 2. In this study, this variant expressed to a higher degree than wild type (Figs. 4B, 6B). Further work will be necessary to understand the reasons for variable degradation in this type of protein engineering.
As for enzymatic activity, specific activity in HeLa cells of ARG1 variant 1 was lower compared with wild type, while specific activity of HPRT1 variant 1 was equivalent to wild type. On the contrary, specific activity in HeLa cells of both ARG1 and HPRT1 variant 2 was higher compared with wild type (Fig. 4E, F), suggesting that some interplay exists between stability and activity that must carefully be considered when designing new variants. Perhaps a more targeted approach using combinations of a few conservative, high-frequency fixation mutations may allow for simultaneous optimization of both thermostability and enzymatic activity [35]. Future work will evaluate this concept.
LNP-formulated mRNA encoding either GFP or HPRT1 or its variants was delivered through i.v. tail injection in wild-type male C57BL/6 mice. Protein expression was analyzed by HRTF assay from liver at 24, 48, 96, and 168 h postdose. For HPRT1 and the variant designed for thermostability, expression was noted at 24, 48, and 96 h with expression peaking at 48 h. Variant 1 expressed at approximately threefold the level of wild type (Fig. 7B) at 48 h, and by 96 h was roughly equivalent to wild type (Fig. 7D). Expression of the variant designed to have higher enzymatic activity peaked at 24 h and dropped off after this point. These results were consistent with the cell-based assays, with the exception of variant 2. Variant 2 expressed to a level similar to wild type in cell-based assays and was relatively unexpressed after 24 h in mouse liver. It is unclear why variant 2 did not express as well as wild-type HPRT1 in vivo, although there may be several reasons. Two possibilities are that the modifications added to the protein sequence introduced proteolytic sites or exposed additional ubiquitinylation sites on the surface. The introduction of either of these changes would result in rapid degradation of the modified protein. Based on these results, the engineering designed to improve thermostability gave a better result for HPRT1. Given that this is a limited study, other targets may yield different results. In fact, a combination of the engineering techniques used in this study may improve results even more. A combination of highly expressed protein with higher activity than wild type may prove to be a powerful therapeutic combination.
In this study, we report that protein engineering can be useful to increase the expression or activity of exogenous protein in cell-based assays or wild-type mice. It is conceivable that protein engineering, coupled with either formulation optimization or mRNA optimization would be useful to improve mRNA therapeutics. Further work will examine whether protein engineering will improve outcomes in animal disease models or whether combination of the protein engineering approaches used in this study could further improve outcome. It is worth noting that the engineering protocols discussed in this study produce several possible sequences that can be examined. In these cases, we examined the top hit from each protocol for ARG1 and HPRT1. Further broadening of the sequences selected and inspection in cell culture may yield even better results. We believe that our protein engineering process, from sequence selection to screening in high-throughput cell-based assays, will continue to yield improved results in vivo and facilitate optimization of mRNA molecules for therapeutic utility.
Footnotes
Acknowledgments
The authors would like to acknowledge the following employees of Alexion Pharmaceuticals, Inc. for their guidance and support: Daniel S. Roseman and Walter Voegtli for protein engineering discussions, Shira Landskroner-Eiger for discussion of statistical analyses, and Christian Michalowski, Kaitlin Losee, Luke Szul, and Joseph Seco for research support.
Funding
This work was supported by Alexion Pharmaceuticals, Inc. Funding for an open access charge: Alexion Pharmaceuticals, Inc.
Author Disclosure Statement
All authors are current or former employees and shareholders of Alexion Pharmaceuticals, Inc., a company that develops therapies for rare and ultra-rare diseases.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.