
Abstract
Since its inception in the early 1990s, SELEX remains the gold standard for discovering RNA aptamers specific for proteins and small molecules. The SELEX process has undergone countless modifications and now encompasses a breadth of innovative selection schemes to pare an aptamer library toward target-specific aptamers. Common to all these RNA aptamer SELEX processes are the steps for the preparation of DNA template and in vitro transcription of aptamer RNA. These steps have remained mostly unchanged over the past three decades and would benefit from optimization. We focused on three key areas: improving the homogeneity of in vitro transcribed aptamer RNA, increasing the efficiency of in vitro transcribed aptamer RNA purification by PAGE, and improving the quality of target-bound aptamer RNA recovered during SELEX. Together, these optimizations contribute toward a more efficient SELEX process and are applicable to both protein-based and cell-based RNA aptamer selections.
Introduction
RNA aptamers and SELEX have come a long way since their beginnings in the 1990s [1,2]. The SELEX process was first reported generating aptamers against recombinant proteins and simple organic molecules [1,2]. Since then, numerous adaptations have been made to the original SELEX protocol, including the identification of aptamers against complex targets, such as cells and whole tissue [3–5]. Detailed protocols have been published with a wide range of innovative strategies employed during the selection of aptamers, both in vitro [6–13] and cell based [3,14–17]. Incorporation of high-throughput sequencing and bioinformatics into the SELEX process has streamlined analyzing large datasets to identify potential candidates [18].
Despite the differences in the selection processes per se and type of RNA aptamer library used, these protocols depend upon a common set of steps, namely double-stranded DNA (dsDNA) template preparation, in vitro RNA aptamer transcription and purification, and recovery of target-bound aptamer RNA during the positive selection steps of SELEX. These steps are rooted to the original SELEX protocol developed in 1990s and have remained mostly unchanged [19]. While not studied often as key parameters for successful aptamer identification, each of these steps greatly influences the quality and quantity of an aptamer library, and thereby contributes to a successful SELEX process. Therefore, the original SELEX protocol needs to be optimized and improvements reported by others in non-aptamer areas of study that make use of transcribed RNA need to be adapted to aptamer-specific protocols [20]. In this study, we report optimizations in three distinct areas; enhancing the 3′-end homogeneity of in vitro transcribed aptamer RNA, improving the yield and decreasing the time necessary for PAGE purification of in vitro transcribed aptamer RNA, and increasing the quality of recovered target-bound RNA aptamers during SELEX.
Methods
A detailed list of materials used and step-by-step description of optimized protocols described in this article are provided under Supplementary Data (Supplementary Data S1—O-Methyl modification of dsDNA for aptamer RNA in vitro transcription.doc; Supplementary Data S2—Purification of in vitro transcribed aptamer RNA.doc; and Supplementary Data S3—Removing aptamer RNA contaminants to improve absorbance 260-230 ratio.doc).
Preparation of dsDNA template for in vitro RNA transcription
Sel2-N20 or Sel2-N25 single-stranded DNA (ssDNA) template oligos were synthesized as unmodified or with 2′-O-methyl-modifications on the 5′-end of the template strand (IDT, Coralville IA) [3].
Unmodified: 5′-TCGGGCGAGTCGTCTG-N20 or N25-CCGCATCGTCCTCCC-3′;
2′-O-methyl modified: 5′-mUmCGGGCGAGTCGTCTG-N20 or N25-CCGCATCGTCCTCCC-3′.
The ssDNA template oligos were extended into dsDNA template by annealing a TATA box containing forward primer (5′-TAATACGACTCACTATAGGGAGGACGATGCGG-3′) and using Taq DNA polymerase [3]. Extended dsDNA template size and purity were confirmed by 4% (w/v) agarose E-Gel EX gels (Invitrogen) and purified using QIAprep 2.0 spin columns (Qiagen).
Transcription of aptamer RNA
RNA aptamers containing modified 2′-fluoro-pyrimidines were in vitro transcribed using either unmodified or 2′-O-methyl-modified template dsDNA at 0.5 μM [3]. The in vitro transcription reactions were carried out in the presence of 1.5 mM NTP mix containing 2′-fluoro-pyrimidines (fUTP and fCTP) and 2′-OH purines (rATP and rGTP) and using the Y693F T7 RNAP [21] at 37°C overnight. Some transcription reactions were supplemented with anhydrous DMSO to a final concentration of 20% (v/v). DNase I was used to digest the dsDNA template post-transcription and the transcribed aptamer RNA was subjected to preliminary purification by phenol-chloroform extraction. Two to five micromoles of transcribed aptamer RNA was separated on 12%–15% (v/v) preparative PAGE gel containing 8M urea. RNA bands were visualized by UV shadowing and the full-length RNA (51 bases for Sel2-N20 and 56 bases for Sel2-N25) excised. Excised aptamer RNA was purified by either using the “Tube-in-a-Tube” or “Crush-and-Soak” aptamer RNA purification methods briefly described below (see Supplementary Data for details). For more sensitive visualization, the polyacrylamide gel containing 100–500 ng aptamer RNA was visualized with SYBR Gold (Invitrogen) and imaged using the Invitrogen E-Gel system. A low range ssRNA ladder (NEB, N0364S) was used to verify the integrity and size of in vitro transcribed aptamer RNA.
Tube-in-a-Tube aptamer RNA purification
Excised gel containing aptamer RNA was placed into a 0.5 mL tube with a hole (20G needle size) at the bottom. The 0.5 mL tube was placed inside a 2.0 mL tube and centrifuged at 20,000 g to force the excised gel through the hole of the 0.5 mL tube into the 2.0 mL tube. Aptamer RNA was eluted from the pulverized gel pieces using the PAGE extraction buffer as described in the Supplementary Data. Yield and quality of purified RNA were determined by Nanodrop (ThermoFisher). A detailed step-by-step protocol of purification of RNA transcripts using the Tube-in-a-Tube method is provided under Supplementary Data (Supplementary Data S2—Purification of in vitro transcribed aptamer RNA.doc).
Crush-and-Soak aptamer RNA purification
Excised gel containing aptamer RNA was placed into a 15-mL conical tube with 4.0 mL TE buffer (containing 10 mM Tris, pH 8.0; 0.1 mM EDTA) and crushed using the end of a 10 mL pipette. RNA was eluted from the crushed gel pieces by passive diffusion for 1–2 h at 37°C. TE containing aptamer RNA was removed and the gel pieces were re-soaked in TE buffer and elution was repeated twice. Eluates were pooled and filtered using a 0.45 μm cellulose acetate syringe filter to remove any gel fragment and transferred to Amicon Ultra centrifugal filter (Ultra 4; 10K MWCO) to concentrate. The concentrated aptamer RNA was washed twice with TE buffer and the quality and quantity of aptamer RNA were assessed by Nanodrop.
Removal of contaminants from recovered target-bound RNA
Trizol-extracted aptamer RNA was filtered to remove common contaminants, including, but not limited to, guanidinium isothiocyanate (GITC) using spin column-based filters. Briefly, Trizol-extracted, ethanol-precipitated aptamer RNA was diluted in polymerase chain reaction (PCR)-grade water and filtered of contaminants using the Amicon Ultra 0.5 mL filters (10K MWCO). Aptamer RNA was washed with additional water and centrifugations, if necessary. The resultant aptamer RNA was assessed for purity and yield by Nanodrop. A detailed protocol is provided in the Supplementary Data (Supplementary Data S3—Removing recovered aptamer RNA contaminants to improve absorbance 260-230 ratio.doc)
Data analysis
All experiments were repeated with at least three biological replicates. Data were analyzed (mean, standard deviation, and standard error of the mean) and plotted using Microsoft Excel and GraphPad Prism software. Statistical significance was determined by Student's t-test and a P value <0.05 was considered significant. A GraphPad Prism file, and pdf of the full report, is provided in Supplementary Data S4 containing metadata and analysis represented by graphs in the figures.
Results and Discussion
Optimization of RNA-based SELEX methods
We identified three distinct areas within the general RNA aptamer SELEX process for optimization to improve the purity and quantity of the aptamer library during SELEX. These optimizations include modifications to the template ssDNA and reverse primer to minimize the 3′-end heterogeneity of aptamer RNA transcripts, PAGE purification of full-length aptamer RNA, and recovery of target-bound aptamer RNA post-selection in the SELEX process. These revised methods are applicable to most RNA-based SELEX processes, from simple protein-based in vitro SELEX [6–13] to more complex whole cell-based and in vivo SELEX [3,14–17].
Increasing the homogeneity of the aptamer transcripts
For library creation in RNA aptamer SELEX, transcription is often carried out using an in vitro runoff assay with the DNA-dependent T7 RNA polymerase (RNAP). Most RNA aptamer SELEX processes utilize a mutant T7 RNAP (Y639F) [21] to incorporate modified nucleotides, such as 2′-fluoro-pyrimidines, within the aptamer RNA to enhance nuclease resistance. During runoff transcription, the T7 RNAP is known to produce shorter transcripts that are abortive products, which are easily removed by size fractionation. However, a sizable population of ‘n+i’ long transcripts is produced by incorporation of one or more non-template nucleotides at the 3′-end of the transcribed aptamer RNA, most frequently adenosines [22–24]. Few studies have investigated the impact of 3′-end heterogeneity on RNA aptamer folding and binding, although artificial addition of a poly-adenylated tail to single aptamers has been investigated [25]. In this study, most of the poly-adenylated aptamers, but not all, retained target binding [25]. However, a direct comparison between the poly-adenylated aptamers and their unmodified counterparts was not available. Thus, the impact of 3′-end heterogeneity on the SELEX process remains unknown, but may be potentially problematic for some aptamer sequences.
Likely, aptamers that are negatively or positively affected by non-templated additions will be at a disadvantage during the SELEX process due to a proportion of the in vitro transcribed aptamer being non-functional. Larger 3′ additions that result in significant size variations will further be eliminated during PAGE-based size fractionation, while smaller additions will often be missed due to the low-resolution preparative PAGE gels. The presence of 3′-heterogeneity of transcribed aptamer RNA may impart an unintended driving factor during SELEX, whereby aptamer sequences resistant to the effect of 3′-end heterogeneity are selected preferentially. Moreover, the length of the aptamer library may be an important factor as shorter aptamer libraries could be impacted to a greater degree by 3′-end heterogeneity than longer aptamer libraries. Overall, the potential effect of 3′-end heterogeneity of transcribed aptamer RNA during the SELEX process should be recognized and strategies, such as discussed in this study, can be included to minimize its occurrence.
The addition of 2′-O-methyl-modified nucleotides into the first two positions of the 5′-end of the template strand of DNA has been reported to significantly reduce 3′-end heterogeneity during in vitro transcription [22,24,26]. Furthermore, the 2′-O-methyl-modified DNA template in concert with 20% DMSO as a cosolvent has also been shown to significantly reduce abortive RNA transcripts during in vitro transcription with 2′OH-NTPs and wild-type T7 RNAP [24,27]. Evaluation of these modifications for transcription of 2′-fluoro-modified RNA aptamers using a mutant T7 RNAP has not been done. We tested independent as well as combined effects of the 2′-O-methyl template modification and the addition of 20% DMSO on reducing 3′-end heterogeneity, abortive transcripts, and overall yield of in vitro transcribed aptamer RNA. The addition of 2′-O-methyl-modified nucleotides to the 5′ end of template strand of the dsDNA is easily achieved by direct chemical synthesis of ssDNA template strand (Fig. 1A) and by incorporating the modification into the first two nucleotides of the reverse primer, which is complementary to the aptamer RNA during cDNA synthesis and to the coding strand of dsDNA during PCR (Fig. 1B). As shown in Fig. 1C and D, conventional in vitro transcription, in the absence of both modifications, resulted in both specific aptamer RNA as well as a prominent larger aptamer RNA transcript. These results suggest that, similar to WT T7 RNAP, the mutant T7 RNAP will incorporate non-templated nucleotides and results in RNA transcripts with 3′-end heterogeneity. The addition of 20% DMSO to transcription with the unmodified template not only decreases the 3′-end heterogeneity but also decreases the overall yield of RNA transcripts (Lane 2). The addition of the 2′-O-methyl nucleotides to the dsDNA template resulted in an increased amount of specific aptamer RNA and reduced levels of the larger transcripts, compared to the unmodified dsDNA template (Lane 3). The addition of 20% DMSO during in vitro transcription of the 2′-O-methyl-modified dsDNA template resulted in predominantly specific aptamer RNA with little to none of the larger aptamer RNA (Lane 4). However, as seen in Lane 2, the addition of DMSO resulted in reduced overall transcript yield. A detailed protocol is provided in Supplementary Data (Supplementary Data S1—O-Methyl modification of dsDNA for aptamer RNA in vitro transcription.doc).
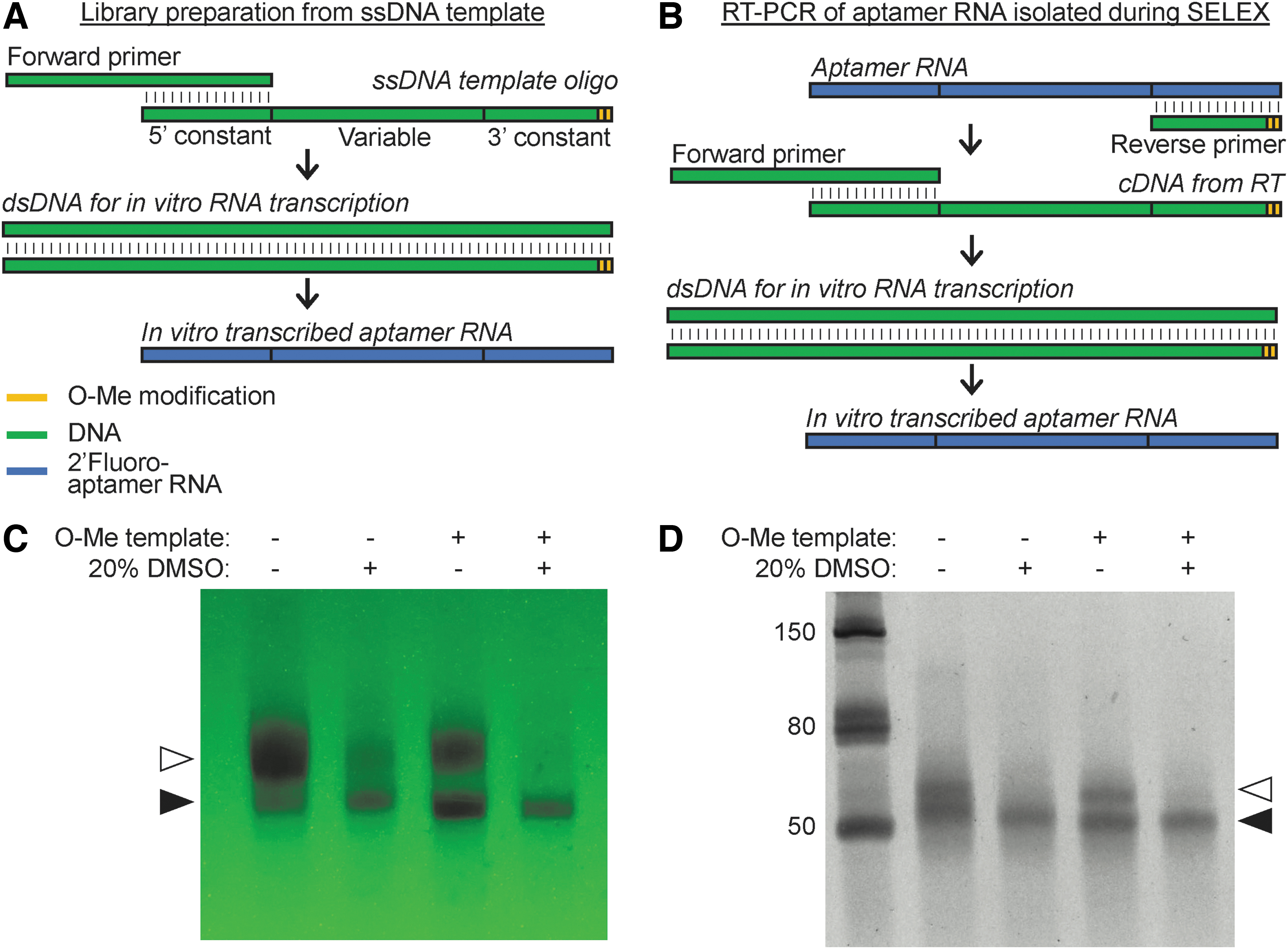
O-methyl modification to dsDNA template to minimize 3′-end heterogeneity. The last two nucleotides of 5′-end of dsDNA template can be modified with 2′-O-methyl (O-Me) nucleotides to minimize 3′-end heterogeneity of in vitro transcribed aptamer RNA. The 2′-O-methyl modifications may be introduced to
Maximizing the gel purification yield of transcribed RNA aptamers
Each round of SELEX can require nanomolar amounts of an aptamer library. Conventionally, an aptamer library is prepared by in vitro transcription reactions using larger volumes (0.5 to 1 mL) followed by preparative PAGE-based purification of the full-length aptamer RNA. Traditional methods used for purifying RNA from these gels result in low elution efficiency. We have optimized the gel extraction and purification methodology through four discrete steps that are faster and more efficient than the current conventional methods. In fact, these gel extraction improvements can mitigate the loss of yield encountered when applying the above described methods to improve 3′-end homogeneity of the transcribed aptamer RNA (Fig. 1).
Phenol-chloroform extraction: we recommend the use of phase-lock gel (PLG) tubes (5PRIME, Quantbio) during the initial phenol-chloroform extraction of the in vitro transcription reactions. PLG tubes greatly facilitate the separation and collection of the aqueous phase and minimize potential accidental contamination by the organic phase.
Sample loading for preparative PAGE: transcription reaction volumes for SELEX aptamer RNA library preparations are typically high enough to require the use of a larger electrophoresis apparatus (eg, Fisher Biotech FB-VE20–1 [3,10] or Labrepco V16–2 [4]). These larger electrophoresis apparatuses are expensive and require difficult-to-handle thin acrylamide gels, and generate more heat during electrophoresis that can result in poor band resolution during UV shadowing. To avoid these complications, we devised a simple method to enable the use of smaller more easily manageable gel systems (eg, BioRad mini protean) for preparative PAGE. The smaller gel systems are limited by the maximum volume that may be loaded into a mini-gel. We overcome this limitation by concentrating the phenol-chloroform extracted aptamer RNA into volumes that can be accommodated by the mini-gels. The phenol-chloroform extracted aptamer RNA is easily concentrated using Amicon Ultra 0.5 mL (10K MWCO) to ∼20–25 μL volume. These lower volumes can be easily loaded onto an 8M urea-TBE-PAGE mini-gel using available comb sizes. Concentrated aptamer RNA is denatured in 3.5M Urea loading buffer (Supplementary Data) containing 6% (w/v) Ficoll 400 (Thermo Fisher). Ficoll 400, which is better than glycerol or sucrose as density gradient media, aids in proper loading of the samples into the bottom of the wells [28–30]. Together, these sample optimizations produce crisper and more easily distinguishable UV-shadowed bands for excision (Fig. 2A).
UV shadowing and excision of aptamer RNA band: UV shadowing the aptamer RNA band is significantly improved by using a Polyolefin gel handler sheet (Z376957; Sigma-Aldrich) placed over the TLC plate. These gel handlers are specifically designed to be UV transparent and are sturdier than the conventional, flimsy plastic wrap, which greatly facilitates the stability of the gel during UV shadowing (Fig. 2A). Moreover, the gel handler provides a smoother and firmer surface than plastic wrap to excise the aptamer RNA bands.
Extraction and purification of excised aptamer RNA band gel fragment: conventionally, RNA aptamers from the excised gel fragment are extracted using a Crush-and-Soak method. The Crush-and-Soak method depends upon passive diffusion of aptamer RNA into a gentler buffer in a concentration-dependent manner. This concentration-dependent process necessitates repeated extractions with larger volumes of the extraction buffer to maintain the concentration gradient and often requires several hours to overnight incubations [3,4,10]. Not surprisingly, the efficiency of Crush-and-Soak method varies widely with between 30% and 60% of the input aptamer RNA. We optimized the aptamer RNA extraction from the excised gel using a Tube-in-a-Tube method (Fig. 2B) [31]. The Tube-in-a-Tube method pulverizes the excised aptamer RNA-containing gel fragment by forcing the gel through a small hole in the bottom of an inner 0.5 mL microfuge tube into an outer 2.0 mL microfuge tube. The pulverized RNA-containing gel pieces are suspended in 1.0 mL of a modified PAGE extraction buffer, containing SDS and sodium acetate, which facilitates the release of the aptamer RNA out of the gel [32]. The gel architecture is further weakened following one or two freeze-thaw cycles, consisting of 10 min at −80°C followed by 10 min at 70°C leading to increased release of RNA. The suspension containing pulverized acrylamide gel is then filtered using a Co-Star Spin-X column. To enable transfer of pulverized gel slurry, we used wide-bore 1.0 mL pipette tips or micropipette tips that have been cut at the tip to provide a larger opening. The filtrate containing the aptamer RNA is then concentrated and washed twice with Buffer TE (containing 0.1 mM EDTA, pH 8.0) using an Amicon Ultra 0.5 mL (10K MWCO) filter. The final preparation of aptamer RNA quality and quantity is assessed by Nanodrop. A single freeze-thaw extraction cycle is enough to recover most of the aptamer RNA. We observed that the Tube-in-a-Tube method resulted in almost twice the extracted aptamer RNA compared to the Crush-and-Soak method with greater consistency between preparations (Fig. 2B). A detailed protocol of this approach is provided in Supplementary Data (Supplemental Data S2—Purification of in vitro transcribed aptamer RNA.doc).
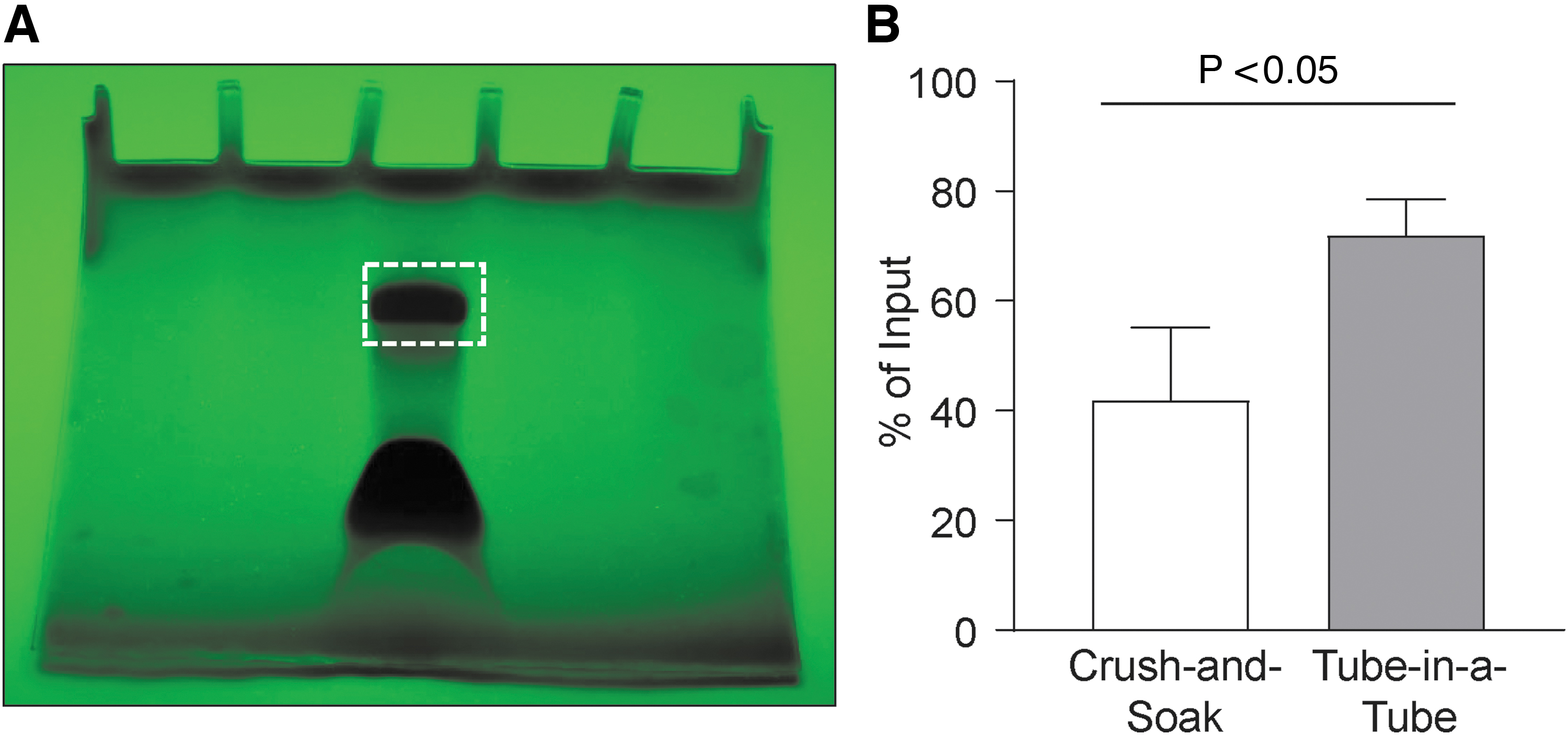
Purification of in vitro transcribed aptamer RNA using optimized methods.
Improving the quality of recovered target-bound RNA aptamers during SELEX
A critical element that determines the success of SELEX proceeding to the next cycle of aptamer library enrichment is the quality of the aptamer RNA recovered from the previous selection cycle. GITC-acid phenol mix, more commonly referred to as TRI-reagent or TRIzol, is frequently used to recover cell-bound or tissue-bound selected aptamer RNA during the SELEX process. GITC is a frequent contaminant carried over to the recovered aptamer RNA, even after ethanol precipitation, and can negatively impact downstream enzymatic reactions, such as reverse transcription and polymerase chain reaction (RT-PCR). Additional ethanol precipitation of recovered RNA will reduce such carryover. However, in addition to the time necessary for ethanol re-precipitation, there is a risk of losing a significant amount of the recovered aptamer RNA with no guarantee that the GITC contamination will have been sufficiently removed. Recovered aptamer RNA with significant GITC contamination exhibits a higher absorbance between 230 and 240 nm, resulting in a lowered OD260/230 ratio (Fig. 3A vs. B). GITC contamination (<100 mM) may not be a practical issue for samples with higher RNA concentrations (>50 ng/mL). However, if the recovered aptamer RNA is very low, as is often the case with aptamer RNA recovered after SELEX, even minor GITC contamination can significantly reduce RT-PCR and quantitative-PCR efficiencies [33–35].
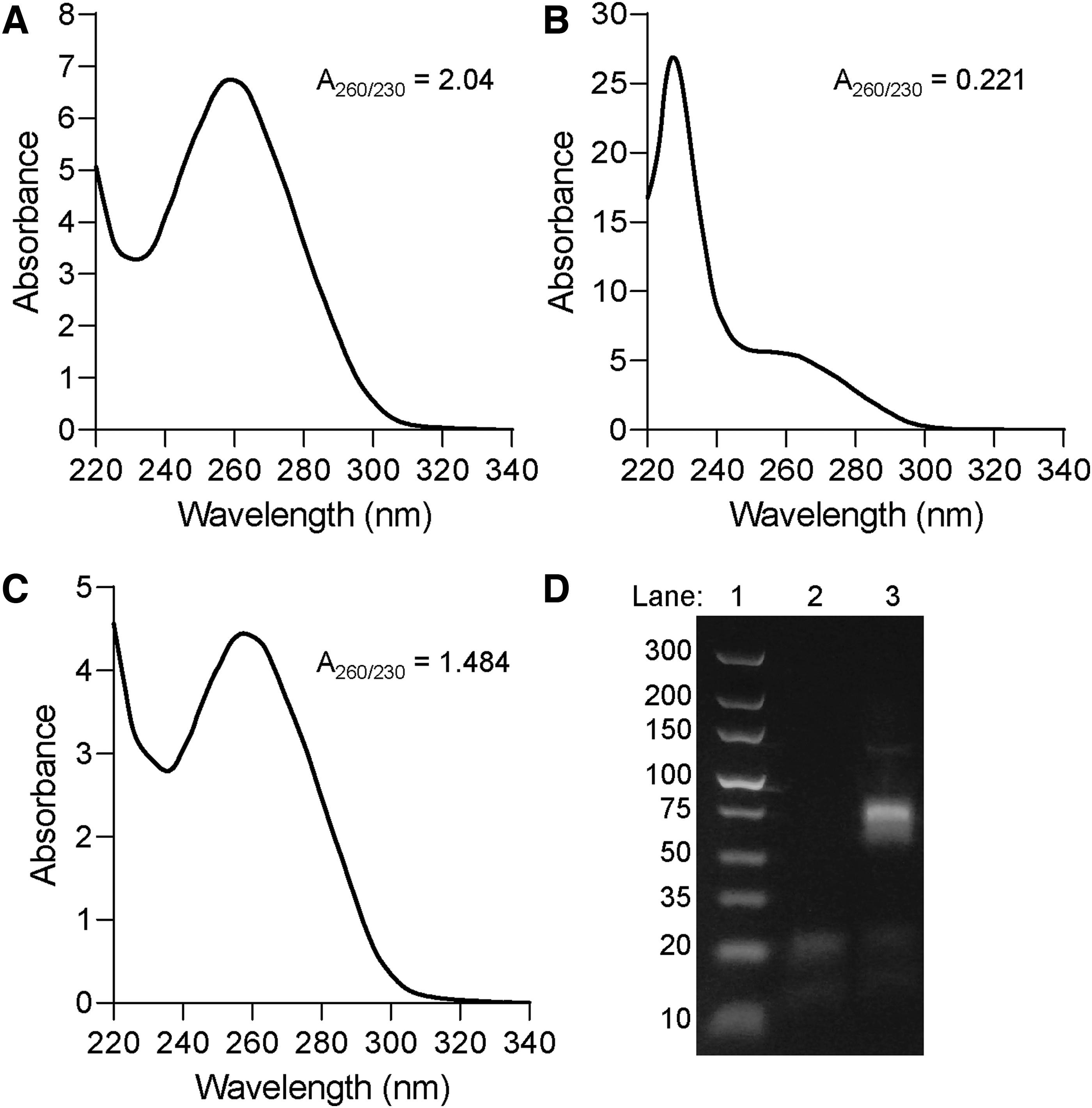
Reducing contamination from TRIzol-extracted aptamer RNA for RT-PCR.
We found that recovered aptamer RNA with significant GITC contamination, as observed by a <1.0 OD260/230 ratio (Fig. 3B), can be easily washed with PCR-grade water using an Amicon Ultra centrifugal filter (10K MWCO). These washes can be conducted quickly and the reduction of GITC contamination can be observed after each subsequent wash by checking the OD260/230 ratio (Fig. 3C). Usually, a single wash is enough to shift the OD260/230 ratio of the recovered aptamer RNA to >1.4, thereby permitting amplification by RT-PCR without interference from GITC contamination (Fig. 3D). A detailed protocol of this approach is provided in Supplementary Data (Supplemental Data S3—Removing recovered aptamer RNA contaminants to improve absorbance 260–230 ratio.doc).
Footnotes
Acknowledgments
The authors would like to thank Dr. George J. Weiner for comments and critiques, while preparing this article and Dr. Kristina Thiel for helpful discussions.
Author Disclosure Statement
The authors have no competing financial interests to disclose related to this work.
Funding Information
This work was supported by the National Institute of Health through the Iowa Mayo Lymphoma SPORE (P50 CA97274), the Holden Comprehensive Cancer Center support grant (P30 CA86862), NIH R01 (HL139581), and by the American Heart Association Innovative Project Award (18IPA34170406).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.