
Abstract
Although MYCN has been considered an undruggable target, MYCN alterations confer poor prognosis in many pediatric and adult cancers. The novel MYCN-specific inhibitor BGA002 is an antigene peptide nucleic acid oligonucleotide covalently bound to a nuclear localization signal peptide. In the present study, we characterized the pharmacokinetics (PK) of BGA002 after single and repeated administration to mice using a novel specific enzyme-linked immunosorbent assay. BGA002 concentrations in plasma showed linear PK, with dose proportional increase across the tested dose levels and similar exposure between male and female and between intravenous and subcutaneous route of administration. Repeated dosing resulted in no accumulation in plasma. Biodistribution up to 7 days after single subcutaneous administration of [14C]-radiolabeled BGA002 showed broad tissues and organ distribution (suggesting a potential capability to reach primary tumor and metastasis in several body sites), with high concentrations in kidney, liver, spleen, lymph nodes, adrenals, and bone marrow. Remarkably, we demonstrated that BGA002 concentrates in tumors after repeated systemic administrations in three mouse models with MYCN amplification (neuroblastoma, rhabdomyosarcoma, and small-cell lung cancer), leading to a significant reduction in tumor weight. Taking into account the available safety profile of BGA002, these data support further evaluation of BGA002 in patients with MYCN-positive tumors.
Introduction
MYCN is a member of the MYC family of oncogenes and is an oncogenic driver in many types of cancers. 1 Deregulation of MYCN occurs in both pediatric and adult cancers. MYCN amplification (MNA) and/or overexpression have been found in pediatric cancers including neuroblastoma (NB), rhabdomyosarcoma (RMS), medulloblastoma, Wilms tumor, and retinoblastoma. In particular, NB is one of the deadliest cancers that occur in early childhood and represents 7% of pediatric malignancies.1–3 About 25% of patients with NB present MNA, which is linked to a poor prognosis, metastasis, and advanced-stage disease.1–3 RMS is the most common pediatric soft-tissue sarcoma and a major cause of cancer death in children. MNA is present in about 25% of cases, and MYCN overexpression occurs in 55% of cases. It is associated with adverse prognosis and is a feature of the more aggressive alveolar subtype (ARMS). 4 Similarly, MNA is also present in adult cancers and occurs in small-cell lung cancers (SCLC) 1 (15%–20%),5,6 neuroendocrine prostate cancers (40%), prostate adenocarcinomas (5%), 7 and basal cell carcinomas (17.5%), 1 whereas overexpression of MYCN is present in a subset of T-cell acute lymphoblastic leukemias, glioblastoma multiforme, and breast cancer. 1 Importantly, the amplification or overexpression of MYCN in the majority of these adult cancers is also associated with a poor prognosis. 1
From a physiological perspective, MYCN expression is restricted during embryogenesis and has a very limited pattern of expression in normal cells after birth. 8 Given its crucial role in MYCN-positive tumors, one should consider this as a promising target. 1 However, N-Myc protein is deemed an undruggable target, and drug discovery approaches aimed at blocking N-Myc protein have largely failed. 9 Although indirect strategies have been proposed, none of these has proven to be effective in standard clinical practice.1,3,10
We have previously demonstrated that an alternative approach consists in the specific gene expression inhibition at the level of chromosomal DNA through MYCN-specific antigene peptide nucleic acid (PNA) oligonucleotide covalently linked to a nuclear localization signal (NLS) peptide.2,4,11–14 The antigene oligonucleotide approach (via persistent and specific block at the level of the target gene transcription) has shown advantages compared with the block of mRNA translation by antisense PNA oligonucleotide strategies.2,4,11–15 In particular, BGA002 is a MYCN-specific antigene PNA that inhibits MYCN gene expression, yielding a potent and specific antitumor activity both in vitro and in vivo.2,13,14 PNAs are oligonucleotide mimetics, in which the anionic sugar-phosphate backbone of the nucleic acids is replaced with an achiral, uncharged polyamide backbone. 16 PNAs showed promising results in diagnostic or therapeutic applications as antisense or antigene drugs owing to their extraordinary stability against enzymatic degradation by proteases and nucleases and their ability to potently and specifically bind with high affinity their target complementary sequences in DNA or RNA.2,4,11,15 PNAs have distinctive characteristics, which allow them to be used for antigene application because their neutral backbone avoids the electrostatic repulsion normally encountered between the negatively charged double-stranded DNA.2,4,11,17 Although PNAs are difficult to synthesize with high efficiency, adding biological function is a more reliable process. In fact, a straightforward approach to improve their pharmacological properties lies in the conjugation of PNA to short synthetic carrier peptides.18,19 Different types of peptides have been evaluated with the aim to improve limited solubility, low cellular uptake, poor biodistribution, and rapid excretion of naked PNA oligonucleotides,2,4,11,18,20–26 which has prevented their broad application as oligonucleotide therapeutics.
In an attempt to explore the pharmacokinetic (PK)–pharmacodynamic (PD) relationship of BGA002 and predict the exposure required for antitumor activity in humans, as well as establish the dosing requirements for a first-time-in-human (FTIH) study, we investigated the PK and PD profiles of BGA002 in pediatric and adult MYCN-positive tumor mouse models after single and repeated doses.27–31 The characterization of the PK in plasma was complemented by the evaluation of the biodistribution in healthy tissues, organs, and blood, taking into accounts the potential effect of route of administration, sex, and accumulation. In addition, to overcome the lack of a standardized methodology, here we report the development and application of a novel hybridization-based enzyme-linked immunosorbent assay (ELISA) approach for the quantification of the MYCN-inhibitor BGA002 PNA-peptide in biofluids, as well as in murine tissues and tumors. We anticipate that, together with information on the safety profile of BGA002, these results provide the basis for the design of oligonucleotide-based cancer therapeutics in a prospective FTIH study. Characterization of the PK and PD of BGA002 may also give a benchmark for the development of other PNA-peptide inhibitors.
Materials and Methods
Chemicals and dose preparation
BGA002 was produced by Biogenera SpA (Bologna, Italy). PNA-peptide was available from stored quantities ready for use or freshly produced by the chemistry department and delivered after purification and dilution for both good laboratory practice (GLP) and non-GLP studies. A solution of BGA002 was quantified through a spectrophotometer (λ260 = 170,300 L/mol/cm) and adjusted to reach 3 mg/mL, then stored at +4°C. Biogenera synthesized an acetylated form of BGA002. The radiolabeled [14C]-Ac-BGA002 (Batch No. 8249DCP006-6, radiochemical purity of 99.8%) was synthesized and supplied by Selcia Ltd (UK) as a solution in acetic acid:sodium acetate:HCl (75 mM:25 mM:3.6 mM) with 2.5% mannitol at a concentration of 1.16 MBq/mL (3.35 mg [14C]-Ac-BGA002/mL). The solution was stored in a fridge set to maintain a temperature of +4°C.
Synthesis of BGA002 compound
BGA002 was synthesized by standard solid-phase peptide synthesis using an automated synthesizer (CEM Liberty Blue instrument with microwave equipment). Solid support is a ChemMatrix resin type, and the strategy employed was 9-Fluorenylmethoxycarbonyl (Fmoc)/Boc. Building blocks employed were protected amino acids (Fmoc and tert-Butyloxycarbonyl/2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl; Boc/Pbf) and PNA monomers (protected as Fmoc/benzhydryloxycarbonyl; Bhoc). Activation strategy employed 4eq of AA/PNA and 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (445460_Sigma Aldrich)/diisopropilethylamine/2,6-lutidine in dimethylformamide (DMF) (for amino acids; AA) or N-methylpyrrolidone (for PNA). Loading of first amino acid was performed in molar defect respect to resin loading with capping (by standard acetic anhydride solution) of the remaining active sites. In this way, resin loading was lowered to ∼0.2 mmol/g (to minimize intrachain interactions during synthesis). After loading of the first amino acid, like in solid phase peptide synthesis (SPPS), the elongation of the oligomer continued by performing iterative synthetic routines for every building block added to the main chain, consisting of:
Deprotection of Fmoc group by reaction with excess 20% piperidine in DMF two times for 5 min. Coupling (single for AA, double for PNA monomers) with the next building block, N,N-Diisopropylethylamine (DIPEA)/2,6-lutidine solution, and HATU as activator at 90°C 170 W for 15 s and 50 W for 110 s for 1 h. Capping by standard Ac2O/2,6-lutidine solution for 15 min.
At the end of the coupling cycles, the compound was then cleaved from the resin as Fmoc derivative by reaction with an 8:2 TFA:m-cresol solution, and after 3 h, the desiderated crude was obtained after precipitation in diethylether. Fmoc-ON purification was required to remove guanidilated compounds. The crude was purified by reverse phase high performance liquid chromatography (RP-HPLC) using a C18 column and standard eluents (water, acetonitrile, trifluoroacetic acid; TFA 0.1%). Unfortunately, the synthesizer cannot perform a preactivation reaction between monomer, base, and activator because only one vessel is present in the instrument. The activator reacts faster with the terminal amine than the monomers and can produce the guanidilate derivative that blocks the sequence. The purified Fmoc compound was dissolved in DMF and a solution of diluted piperidine was slowly added for the deprotection reaction. The corresponding crude material obtained was purified again using the same instrument and conditions, obtaining the BGA002 as a TFA salt.
Final steps of BGA002 production consist in counter-ion exchange (trifluoroacetate to chloride) and lyophilization. TFA counterion could be exchanged both by basic precipitation and aqueous HCl dissolution or by chromatography. Yield of the whole process was ∼5%.
Labeling Ac-BGA002 with carbon-14
The labeling of [14C]-Ac-BGA002 has been performed in accordance with the Selcia Quality Program by the Radiochemistry group at Selcia Ltd. Two grams of resin-bound PNA oligomer Fmoc protected was placed into the SPPS reactor; the resin was rinsed with DMF and then covered with DMF and left to swell for 2 h. The remaining DMF was flushed away with nitrogen, and the resin was exposed to the deprotection solution (20% of piperidine in DMF) and then shook for 20 min at room temperature for two times. A Kaiser test was performed on a small amount of resin to confirm that deprotection of the amino functionality had occurred. [1-14C]sodium acetate was azeotroped with toluene and suspended in DMF and lutidine and DIPEA were added. The resulting mixture was stirred at room temperature under nitrogen until a clear solution was obtained (around 10 min). The resulting orange solution was then added to the SPPS reactor and showed that the sampling had not gone to completion. The solution was flushed away using nitrogen and the resin was rinsed with DMF. Another equivalent of [1-14C]sodium acetate was activated using HATU, 2,6-lutidine, and DIPEA in DMF. The activated mixture was added to the SPPS reactor and shaken overnight at room temperature; unfortunately, the Kaiser test showed that the coupling had not gone to completion; therefore, the mixture was given more time to react and was stirred at room temperature for another 5 days. A Kaiser test was performed on a small amount of resin at the end of each coupling reaction to check the completion of reaction itself. Another equivalent of [1-14C]-sodium acetate was activated using HATU, 2,6-lutidinide, and DIPEA in DMF, and the resulting mixture was transferred into the SPPS reactors. The mixture was shaken overnight at room temperature. The Kaiser test performed showed that the coupling had not gone to completion; therefore, the mixture was given more time to react and was stirred at room temperature for another 24 h. The solution was flushed away using nitrogen, and the resin was rinsed with DMF followed by dichloromethane (DCM) and diethyl ether. This rinsing sequence was repeated another three times. The resin was covered with the cleaving solution, and the SPPS reactors were placed on the shaker for 3 h. The reaction mixtures were discharged in Falcon tubes containing ethyl ether. This caused a precipitate to form. The resin was washed with TFA, and the rinsing was collected into a Falcon tube. The tubes were stored at −15°C for 2 h and then centrifuged at 4,000 rpm for 15 min. The supernatant was removed, and the resulting solid deposit was further washed with diethyl ether using the centrifuge. The Falcon tubes were stored in a fridge at −4°C overnight. The solid residues at the bottom of the Falcon tubes were dried under a stream of nitrogen for 2 h and then over a stream of phosphorus pentoxide in a desiccator for another 2 h. The crude was purified by RP-HPLC using a C18 column and standard eluents (water, acetonitrile, and TFA 0.1%).
Animals: Mice
NOD/SCID CB17 mice for PK analysis in tumor and liver and for efficacy studies were born in the animal facility of the Department of Veterinary Medical Science of the University of Bologna (Italy). All experiments were approved by the Scientific Ethical Committee of Bologna University according to Protocol No. 7/73/2012 and authorization 564/2018-PR. PK plasma analysis was performed in juvenile mice (CD1-Swiss, 5 weeks old) by Glp Life Test (Bologna, Italy). Aptuit (Verona, Italy) and Farefarma (Novara, Italy) performed, respectively, ELISA analysis and PK analysis. Biodistribution analysis in mice (CD1-Swiss, 5 weeks of age) was performed by Charles River (Tranent, Edinburgh). Animals were handled according to internal standard operating procedures. PK and biodistribution studies in mice were performed under GLP compliance.
In vivo PK/TK experiments in mouse plasma
The animals used for the studies included Swiss mice (CD1) (5 weeks old at start of treatment) from the Charles River Laboratories Italy Srl with a health certificate according to Legislative Decree 24/20142.
The rationale for the dose selection of the single-dose PK study in mice was based on feasibility studies performed by Biogenera, which showed detectable concentrations at 15 mg/kg dose in tumor and liver; thus, this dose and the threefold lower dose 5 mg/kg were selected to obtain data in plasma. The rationale for the dose selected for the repeated-dose PK studies in mice was based on the results from the single-dose PK study. Given that doses of 5 and 15 mg/kg resulted in distinct concentration versus time profiles, the PK/toxicokinetics (TK) study included doses of 2.5, 7.5, and 25 mg/kg/day, under the assumption of linearity across the dose range.
For the single-dose study, 24 male and 24 female mice for each dose level and route of administration, that is, intravenous (bolus) injection (IV) through the caudal vein or subcutaneous injection (SC) on the back, were treated with 100 µL of a solution containing BGA002. In addition, for each route of administration, a control group of four males and four females was treated with 100 µL of the vehicle. In total, 104 mice received BGA intravenously and 104 mice received the drug subcutaneously. The animals were sacrificed at six different time points after dose for blood collection (time points IV: 5–15–30–60–240–480 min; time points SC: 10–30–60–90–180–480 min).
For the repeated-dose study, 36 female mice for each dose of BGA002 were treated by SC injection with a constant injection volume of 5 mL/kg/day BGA002. The volume of solution administered was calculated weekly using the last mean body weight for the group. Eighteen animals per group were treated with a single injection, whereas another 18 animals per group were treated for 28 days with a once-a-day SC injection. In total, 108 female mice were allocated to six different treatment groups. The animals were sacrificed at six different time points after dose (10–30–60–90–180–480 min) for blood collection, with three animals per time point.
In both studies, the blood was collected from the vena cava under anesthesia by inhalation of a mixture of CO2/O2 (70% and 30%, respectively). In total, 0.3 mL of whole blood was drawn from each animal per time point and, owing to technical limitations in juvenile animals (5 weeks old at start of treatment), the samples were pooled in a single tube containing lithium-heparin (APTACA mod 2400/1). The plasma collected from each tube was frozen at about −20°C overnight and shipped on dry ice for analysis.
Hybridization-based ELISA in plasma and organs (tumor/liver)
A hybridization-based ELISA has been developed and validated to measure the concentration of BGA002 present in plasma samples and in tumor/liver samples. This assay is an attractive bioanalytical tool owing to its ultrasensitivity, microscale sample volume, and ease of use with little or no sample cleanup, which make it a suitable tool to study the TK/PK profiles of therapeutic oligonucleotides to support preclinical and clinical development programs.
Neutravidin-coated plates were incubated with a capture probe, biotin-labeled, and partially complementary to BGA002. After incubation and washing, plasma samples were added to the plate where the complex was immobilized by the neutravidin-biotin binding. After washing, a digoxigenin-labeled detection probe complementary to the other part of BGA002 was added. Then a detection solution containing anti-digoxigenin-peroxidase (anti-DIG-POD) was added, making possible visualization by addition of tetramethylbenzidine (TMB), which produces a colored product. The amount of color correlates to the amount of BGA002 present. More in detail, plates were washed three times with 300 µL of 5X SSCT buffer; 100 µL of 0.2 µM capture probe solution in 5X SSCT buffer was added and incubated at room temperature (RT) for 30 min. Plates were washed three times with 300 µL of 2X SSCT buffer; 100 µL of plasma samples/control sample/quality control was added and incubated at 37°C for 60 min. Plates were washed four times with 300 µL of 2X SSCT buffer; 100 µL of 0.2 µM of detection probe solution in 5X SSCT buffer was added and incubated at RT for 60 min. Plates were washed three times with 300 µL of 2X SSCT buffer and three times with 300 µL of purified water; 100 µL of anti-DIG-POD Fab fragments (1:8,000) in phosphate-buffered saline with Tween and incubated at RT for 60 min. Plates were washed three times with 300 µL of 2X SSCT buffer; 100 µL of substrate solution (1-step Ultra TMB ELISA) was added and incubated at RT for a period of 15 min. Reaction was stopped by adding 100 µL of 0.5 M sulfuric acid. Reading was performed on Spectra Max 250 at 450 nm with reference at 750 nm.
Validation of hybridization-based ELISA
The hybridization-based ELISA described above was validated in mouse tumor and liver for GLP-like use and in mouse plasma and rabbit plasma for GLP use as follows:
Validation in mouse tumor and liver homogenate was performed over the range 200–3200 ng/mL BGA002 and 100–3,200 ng/mL BGA002, respectively. Calibration standards were prepared from a set of working solutions at 10 concentrations of the test item plus blank in the appropriate matrix, yielding concentrations between 15.63 and 8,000 ng/mL BGA002 (Fig. 1b). Validation samples to establish the accuracy and precision of the method were prepared from test item aliquot at nominal concentrations of 50, 100, 200, 400, 800, 1,600, and 3,200 ng/mL BGA002. The concentration for the dilution quality control (DQC) tested in the linear range was 80,000, 40,000, 20,000, and 10,000 ng/mL.
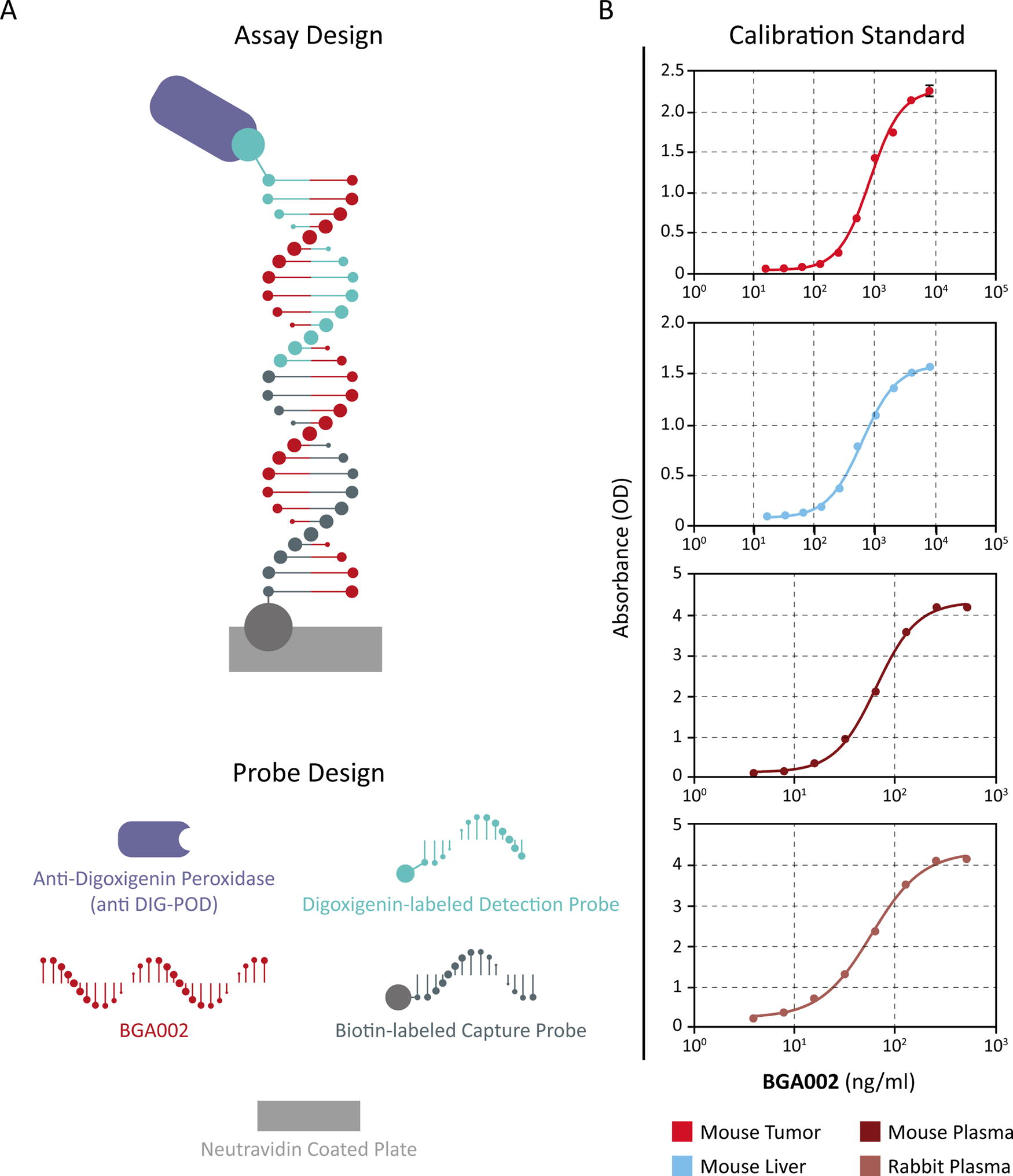
Hybridization-based ELISA approach for the quantification of BGA002.
Validation in mouse plasma has been performed over the range 20–200 ng/mL. Calibration standards were prepared from a set of working solutions at eight concentrations of the test item plus blank in the appropriate matrix, yielding concentrations between 3.91 and 500 ng/mL BGA002. Validation samples to establish the accuracy and precision of the method were prepared from test item aliquot at nominal concentrations of 20, 50, 100, 150, and 200 ng/mL BGA002. The plasma concentration for the DQC tested in the linear range was 20,000 ng/mL.
The following parameters were calculated: lower limit of quantification (LLOQ), upper limit of quantification (ULOQ), precision (% coefficient of variation; CV) intraassay, precision (%CV) interassay, accuracy (%bias), selectivity interference with the analyte from individual lots of plasma, specificity, stability (at room temperature, +4°C, −20°C), freeze–thaw stability, and prozone effect.
Pharmacokinetic analysis
PK analysis was performed using a noncompartmental approach in Phoenix Win-Nonlin 6.3 (Pharsight Corporation, USA). All concentration data points were weighted by the inverse square of the fitted value. The following parameters were evaluated: time to reach maximum concentration (Tmax), peak concentration (Cmax), area under the concentration versus time curve (AUC), elimination half-life (t1/2 elim), volume of distribution (Vd), total clearance (Clb), and bioavailability (F%).
Peak concentration (Cmax) and the time to reach Cmax (Tmax) after administration of the BGA002 were determined from the observed data. The AUC was calculated using the linear trapezoidal rule (calculation method: linear trapezoidal with linear interpolation). The first-order constant (λz) associated with the terminal (log-linear) portion of the curve was estimated by linear regression of the time versus log concentration, and the terminal half-life was calculated as ln(2)/λz. The volume of distribution (Vd) was calculated based on the terminal phase as Dose/λz·AUCinf_obs (where AUCinf_obs is the AUC from the time of dosing extrapolated to infinity). The total clearance (Clb) was calculated as Dose/AUCinf_obs. The bioavailability (F) after subcutaneous administration was calculated as the ratio of the AUC after subcutaneous (AUCsc) and intravenous (AUCiv) administration as (AUCsc/AUCiv) multiplied by 100.
Biodistribution of BGA002 in mice (QWBA-Charles River/Selcia)
The [14C]-Ac-BGA002 (Batch No. 8249DCP006-6, radiochemical purity 99.8%) was supplied by Selcia Ltd (UK) as a solution in acetic acid:sodium acetate:HCl (75 mM:25 mM:3.6 mM) with 2.5% mannitol at a concentration of 1.16 MBq/mL (3.35 mg [14C]-Ac-BGA002/mL) and was stored at +4°C. CD-1 mice (28–35 days of age) were supplied by Charles River, UK, and acclimatized to the experimental unit for 7 days prior to use in the study. The [14C]-Ac-BGA002 formulation was administered by injection into the nape of the neck (single subcutaneous administration) of six male and six female mice at a target volume of 4.5 mL/kg to achieve a nominal dose level of 15 mg/kg (target radioactive dose of 5 MBq/kg). One male mouse and one female mouse were euthanized by CO2 narcosis after 1, 4, 8, 24, 48, and 168 h post dose.
The carcass of each animal was then frozen by immersion in a mixture of solid CO2 in hexane for approximately 15 min and embedded in a block of carboxymethylcellulose. After equilibration at −20°C, sagittal sections (30 µm thick) were taken through each animal using a whole-body cryomicrotome (Leica Instruments GmbH). The sections were freeze-dried before storage at ambient temperature. All samples prepared in scintillation fluid were subjected to liquid scintillation counting for 5 min, together with representative blank samples using a liquid scintillation analyzer with automatic quench correction by an external method. Where possible, samples were analyzed in duplicate and allowed to heat and light stabilize before analysis. Before the calculation of each result, a background count rate was determined and subtracted from each sample count rate. For scintillation counting, a limit of reliable measurement (LRM) of 30 c.p.m. above background was introduced in these laboratories. Where results have arisen from data below the LRM, the fact was noted. The radioactivity present in various organs and tissues in whole-body sections was determined by quantitative whole-body autoradiography (QWBA) using a Typhoon FLA7000 scanner and AIDA image analysis software (version 4.06, raytest isotopenmeβgerate GmbH, Germany). For analysis, representative whole-body sections were placed into close contact with phosphor screens and left for a period of 7 days. On each phosphor screen, a set of external standards was also exposed. These standards were prepared from blood spiked with a serial dilution of a [14C]-labeled reference solution, which was dispensed into holes drilled into a block of carboxymethylcellulose, frozen, and then sectioned in the same way as the animal samples. After the phosphor screen was scanned, an image of the radioactivity in the sample was stored digitally. For quantitative analysis, six background areas were defined on each storage phosphor screen image. The software automatically calculated the mean background and subsequently subtracted this from all standards and tissues analyzed. A regression coefficient was derived by comparing the response of each standard with the nominal concentration over the range of radioactive concentrations used and forcing the response curve through the origin. The concentrations of the standards used were in the range of 0.05–258.48 µg equiv/g. The response curve was linear over these concentrations and assumed to be linear to the limit of reliable determination. Each organ or tissue of interest was then identified and integrated, and the software automatically calculated the concentration (µg equiv/g) using the regression equation derived from the standards.
The LRM for each storage screen was calculated from the assessment of the mean background of the plate and defined as three times the standard deviation. At the specific activity used in this study, the LRM was in the range of 0.01–0.04 µg equiv/g.
Xenograft mouse models
Luminescent cells (Kelly-Luc, H69AR-Luc, RMZ-Luc) and xenograft ectopic mouse models (NB, SCLC, RMS) were prepared according to previously published studies. 2 Kelly cell line was originally established from a 1-year-old female patient with a stage IV NB with MYCN amplification, purchased from DSMZ (DSMZ Cat# ACC-355, RRID:CVCL_2092). The alveolar RMS cell line RMZ was kindly provided by Prof. Lollini (Department of Experimental Pathology, University of Bologna) and was characterized by MYCN amplification. 32 The SCLC H69AR cell line was originally established from the pleural fluid of a 55-year-old Caucasian male with small-cell carcinoma of the lung, purchased from ATCC (CRL-11351). The H69AR cell line is a multidrug-resistant cell line as compared with the parental NCI-H69 cell line. The H69AR cell line presents MYCN gene amplification and high expression of MYCN mRNA and protein.33,34
Pharmacokinetics evaluation of BGA002 in tumor and liver of NB, RMS, and SCLC xenograft mouse models
NB, SCLC, and RMS mouse models were established as described above. In each mouse model, systemic treatment with BGA002 was administered subcutaneously and started when specific bioluminescent value (in NB and SCLC mouse model) or tumor volume (in RMS mouse model) was reached. NB mice were treated with BGA002 daily at 10 mg/kg/day (tumor n = 4; liver n = 7) or vehicle (n = 5) for 15 consecutively days, SCLC mice were treated with BGA002 at 15 mg/kg/day (tumor n = 13; liver n = 13) or vehicle (n = 7) for 9 consecutive days, whereas RMS mice were treated with BGA002 at 15 mg/kg/day (tumor n = 3; liver n = 3) or vehicle (n = 5) for 15 days. Tumors and livers were extracted immediately after animal euthanasia, weighted on a precision scale, and homogenized with a probe sonicator in RIPA extraction buffer at a concentration of 10 mg/mL. Concentrations of BGA002 in tumor and liver were evaluated by the hybridization-based ELISA as described above. Samples were stored at −20°C.
Pharmacodynamics of BGA002 in RMS and SCLC mouse models
BGA002 or vehicle was administered subcutaneously to mice in the SCLC and RMS tumor models, respectively, at doses of 15 mg/kg/day for (n = 4)/vehicle (n = 5) or 5 mg/kg/day (n = 4)/vehicle (n = 3) for 14 consecutive days. In both cases, treatment started when a predefined tumor volume (in RMS mouse model), or bioluminescent value (in SCLC mouse model) was reached. Twenty-four hours after the last injection, animals were euthanized, and necropsy was performed.
Tumors were extracted and weighted on a precision scale and all organs collected for subsequent analysis and studies. Western blot production of N-Myc protein was assessed in total protein extracts. Sample was removed from the animal as quickly as possible after animal sacrifice and homogenized with a probe sonicator on ice in sample lysing solution added with a Halt protease inhibitor cocktail (Cat# 78429, Thermo Fisher) in a 1:100 rate. Sample lysing solution volume should be at least 200 μL and at most 2 mL and adjusted to a concentration of 100 mg/mL. Total protein extract was quantified with the bicinchoninic acid method at NanoDrop ND-1000 spectrophotometer against a standard curve of bovine serum albumin (BSA) in sample lysing solution. Proteins were separated by one-dimensional precast polyacrylamide gel (Bolt Bis-Tris Plus Gels 10%) that provides a neutral pH environment with the aim to minimize protein modifications under denaturing conditions. Samples containing proteins were denatured by incubation at 95°C for 10 min after the addition of 4X sample buffer, 10X reducing agent, and water to a final volume of 30 µL. Each lane was loaded with 30 µg of protein and run at 200 V for about 15 min in electrophoresis buffer (Bolt MES SDS Running buffer) with the molecular weight marker SeeBlue Plus2 Pre-Stained Standard. They were transferred from gel to polivinilidenfluoruro membrane (IBlot Transfer Stack) using the IBlot Gel Transfer Device, a dry system that transfers proteins in 7 min (all materials and reagents were purchased from Life Technologies). The membrane was subjected to overnight incubation at 4°C with blocking solution (5% milk in PBS-Tween 0.1%). The day after it was incubated for 1 h with the primary antibody, a mouse monoclonal anti-N-Myc antibody (SC-53993_Santa Cruz) diluted 1:200 in 3.5% BSA in PBS-Tween 0.1%. After three washes in PBS-Tween 0.1%, the membrane was incubated for 1 h with the corresponding secondary antibody, a sheep anti-mouse IgG-HRP (Amersham Biosciences) diluted 1:10,000 in 3.5% BSA in PBS-Tween 0.1%. The membrane was washed three times in PBS-Tween 0.1% for 5 min before detection. N-Myc chemiluminescent signal was measured adding the ECL Select Western Blotting solution (GE Healthcare) at ChemiDoc-It (UVP) to the membrane. The signal quantification was carried out with Alliance software (Uvitec) and was performed following colloidal staining with Coomassie brilliant blue dyes. All N-Myc protein signals were normalized using the Coomassie brilliant blue signal for each lane.
Immunohistochemistry (IHC) analysis was performed on formaldehyde-fixed and paraffin-embedded samples, cut on the microtome. Antigen retrieval was performed with EDTA-NaOH pH 8. Antibodies used were Anti-MYCN primary antibody (OP13 Calbiochem) and HRP-conjugated secondary antibody (Dako). Staining was performed with the DAKO kit for horseradish peroxidase (HRP) colorimetric revelation using 3,3′-Diaminobenzidine and hematoxylin contrast.
Statistical analyses
All statistical analyses were performed using GraphPad Prism software (GraphPad Software Inc.) version 6. Data from distinct groups were compared using the Mann–Whitney test. Statistical significance level was set at P < 0.05.
Results
Validation of the hybridization-based ELISA approach for the quantification of the MYCN-inhibitor BGA002 PNA-peptide in biofluids and tissues
Hybridization-based ELISAs have been used for the determination of different kinds of oligonucleotide (ODN), including phosphorothioate antisense oligonucleotide,35–37 mRNA, 38 modified oligonucleotides including PNA and 2’-O-MOE PS, 18 siRNA, and unmodified DNA ODN. 35 We set up the method based on the assay developed by Straarup et al. 38 having a sandwich structure with BGA002 that links together the capture and detection probes, specifically binding these two key elements (Fig. 1a). The general sensitivity of the assay is very high; LLOQ in plasma reaches the value of 20 ng/mL and of 200 ng/mL in tissue homogenate (Fig. 1b and Table 1). Also the precision is very high, considering that the interassay precision and intraassay precision are always under about 10%CV (Fig. 1b and Table 1). In contrast, accuracy shows larger differences between liver tissue and tumor, with bias ranging between −29.0% and 19.6% for mouse liver and between –15.7% and 2.4% for mouse tumor (Fig. 1b and Table 1). For GLP study only, we also evaluated the stability of the assay. System stability at −20°C (nominal) was high, with the system maintaining its characteristics up to 3 weeks. At room temperature, the stability is at least 4 h. Also the freeze–thaw stability is the same and is settled for at least three cycles from −20°C (nominal) to room temperature. No prozone effect has been observed. Overall, our results demonstrated that this method is specific for the intended analyte, BGA002, and is able to quantify it in different complex matrices. These results confirm that the hybridization-based ELISA is an appropriate tool to obtain oligonucleotide TK/PK profiles in preclinical and clinical drug development.
Hybridization-Based ELISA Parameters in Mouse and Rabbit Tissue Homogenate
Table reporting complete hybridization-based ELISA parameters calculated in mice and rabbits tissue homogenates. In order are reported data for the four different matrixes used in the validation. The table covers both GLP-like and GLP data.
Abbreviations: ELISA, enzyme-linked immunosorbent assay; LLOQ, lower limit of quantification; ULOQ, upper limit of quantification.
Pharmacokinetics of BGA002 in mice
The PK profiles of BGA002 were comparable after single dose and after 28 days of repeated SC administration, as demonstrated by both Cmax and AUC(0-t) values. Absorption following SC administration was fast, with Tmax being achieved after 10 min. Systemic exposure was similar between male and female, irrespective of the dose level, with similar exposure between IV and SC administration (Fig. 2a, b, and c, Supplementary Fig. S1, Supplementary Fig. S2, and Supplementary Fig. S3). In addition, exposure increased with increasing doses, despite the lack of evidence for dose proportionality (Fig. 2e, Supplementary Table S3). BGA002 was quantifiable up to 8 h in plasma (Fig. 2a, b, and c, Supplementary Table S1 and S2), after which drug levels were below the limited of detection. Despite interindividual variability, none of the doses showed accumulation after repeated dosing (Fig. 2d, Supplementary Table S3). The AUC(0-t) after single SC administration of 2.5, 7.5, and 25 mg/kg dose was 478.1, 1331.1, and 3433.6 h*ng/mL, respectively. A similar exposure range was observed after repeated dosing, namely, 428.8, 1208.7, and 2801.5 h*ng/mL after 2.5, 7.5, and 25 mg/kg dose, respectively (Table 2). Likewise, Cmax also increased in a dose-dependent manner (198, 410, and 1031 ng/mL after 2.5, 7.5, and 25 mg/kg dose, respectively). Volume of distribution (Vd) values suggest a high distribution into tissues, with potential tissue uptake and accumulation, even though plasma levels remain comparable after repeated dosing. The elimination half-life (t1/2) was in fact generally short, in line with the estimates of systemic clearance (Clb). The median of relative bioavailability (F) for the subcutaneous route, independently from the dose, was calculated as 98.4% in male and 83.6% in female mice (Table 2).
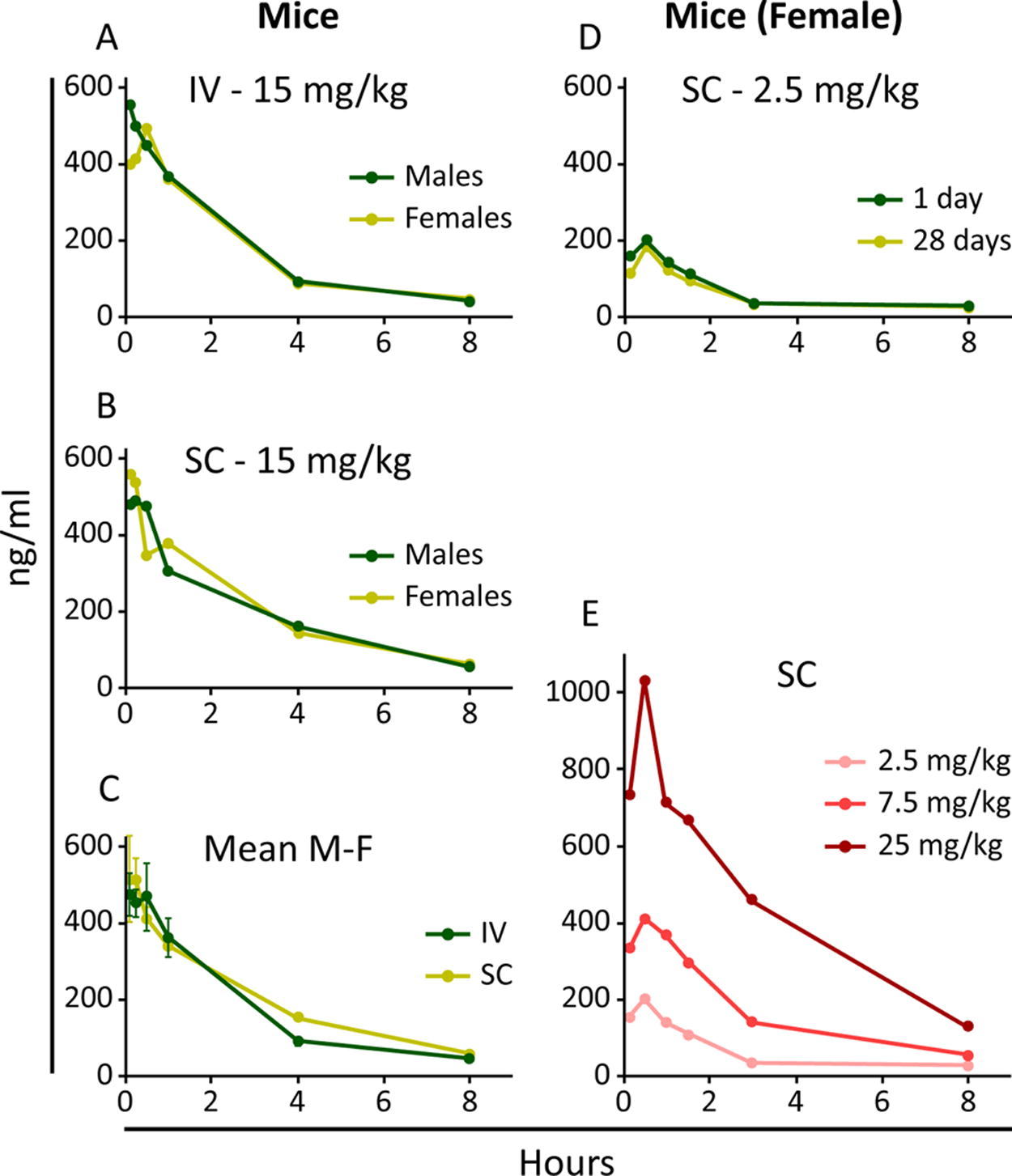
BGA002 pharmacokinetics in mice.
Pharmacokinetics Parameters in Mouse
Table reporting complete PK parameters calculated in mice. In order are reported data after subcutaneous (SC) or intravenous (IV) single-dose administration in mouse and single vs. repeated subcutaneous (SC) administration in mouse. Each estimate is reported as mean of multiple single values.
Abbreviation: AUC0-t, area under the concentration vs. time curve; AUCinf, AUC from the time of dosing extrapolated to infinity; Clb, total body clearance; Cmax, peak concentration; F, bioavailability; Tmax, time to reach maximum concentration; t1/2 elim, elimination half-time; Vd, volume of distribution.
BGA002 has a broad biodistribution in organs and tissues in mice
In order to examine the tissue biodistribution of BGA002 after single subcutaneous administration, radioactive BGA002 containing a [14C]-acetylated at the C-terminus ([14C]-Ac-BGA002) was injected into male and female CD-1 mice at a target dose of 15 mg/kg. Total radioactivity in selected tissues was investigated by QWBA at several time points (1, 4, 8, 24, 48, and 168 h post dose) (Fig. 3). The total radioactivity in selected organs and tissues of male CD-1 mice is presented in Figure 3a and 3b with a representative example of the autoradiography shown in Figure 3c. The results indicate a wide distribution of BGA002 into several tissues and organs, with the kidney, liver, spleen, bone marrow, lymph nodes, and adrenals (Fig. 3a and b) exposed to the highest levels of radioactivity. The results also confirmed fast clearance from blood, with the highest concentrations of BGA002 detected at the first sampling time point (i.e., 1 h post dose, 1.20 µg equiv/g) and less than half the levels at 4 h post dose, with further decrease in the subsequent time points. The majority of the tissues showed high concentration between 4 and 24 h post dose, but it was not possible to identify a clear Tmax. In both sexes, diffusion and biodistribution from the dose injection site was very slow, with radioactivity still visible at the injection site and in the surrounding areas at 168 h post dose (last time point) (Fig. 3a and b). Radioactivity remained well above the limit of detection in most tissues at this time point. The elimination half-life could not be estimated owing to the high level of radioactivity still present at the last time point. Radioactivity levels in the spinal cord and brain in both male and female animals were low, indicating limited penetration through the blood–brain barrier (Fig. 3a, b, and c).
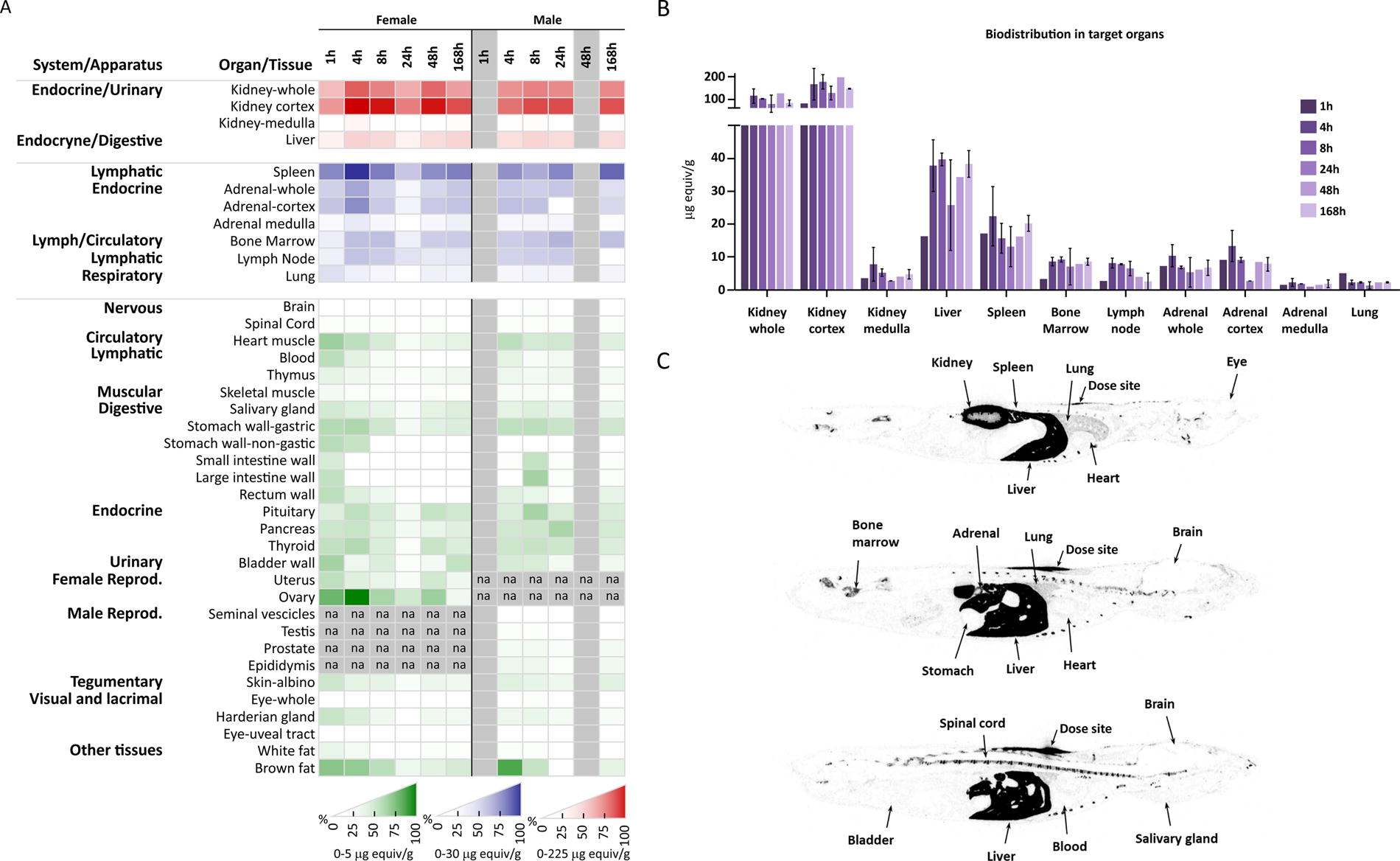
BGA002 biodistribution in male and female mice after single SC administration.
BGA002 highly concentrates in tumors and correlates with efficacy in different xenograft mouse models
The PK profile of BGA002 in tumors was investigated in three tumor xenograft mouse models with MNA and MYCN-expression (NB, RMS, and SCLC). Our results reveal that BGA002 can be found into the tumors in all the three mouse models (Fig. 4 and Table S6). In particular, in NB tumors, BGA002 reached a concentration threefold higher compared with the liver (Fig. 4 and Table S7). The antitumor activity of BGA002 was evaluated in vivo in the same three xenograft murine models. In the RMS model, treatment with BGA002 (SC administration of 15 mg/kg/day for 15 days) resulted in tumor weight decrease by more than 70% (Fig. 5b). In addition, BGA002 (SC administration of 5 and 15 mg/kg/day for 9 days) induced tumor weight decrease in the multidrug-resistant SCLC mouse model of 15% and 59% (P < 0.05), respectively (Fig. 5b), highlighting agreement with previous data. 13 Similarly, reduction of the N-Myc protein was observed in the RMS model and multidrug-resistant SCLC mouse (Fig. 5a and c). In this regard, we intentionally decided to use IHC analysis for qualitative evaluation of the effect of BGA002 on protein. Interestingly, this trend of reduction is observed in Western blot in both SCLC and RMS model (Fig. 5b). However, statistical significance appears only in RMS (P < 0.05). Finally, a similar pattern of response was observed in a histological analysis of tumor vascularization, leading to tumor vessels elimination after treatment at 15 mg/kg/day (Supplementary Fig. S4).
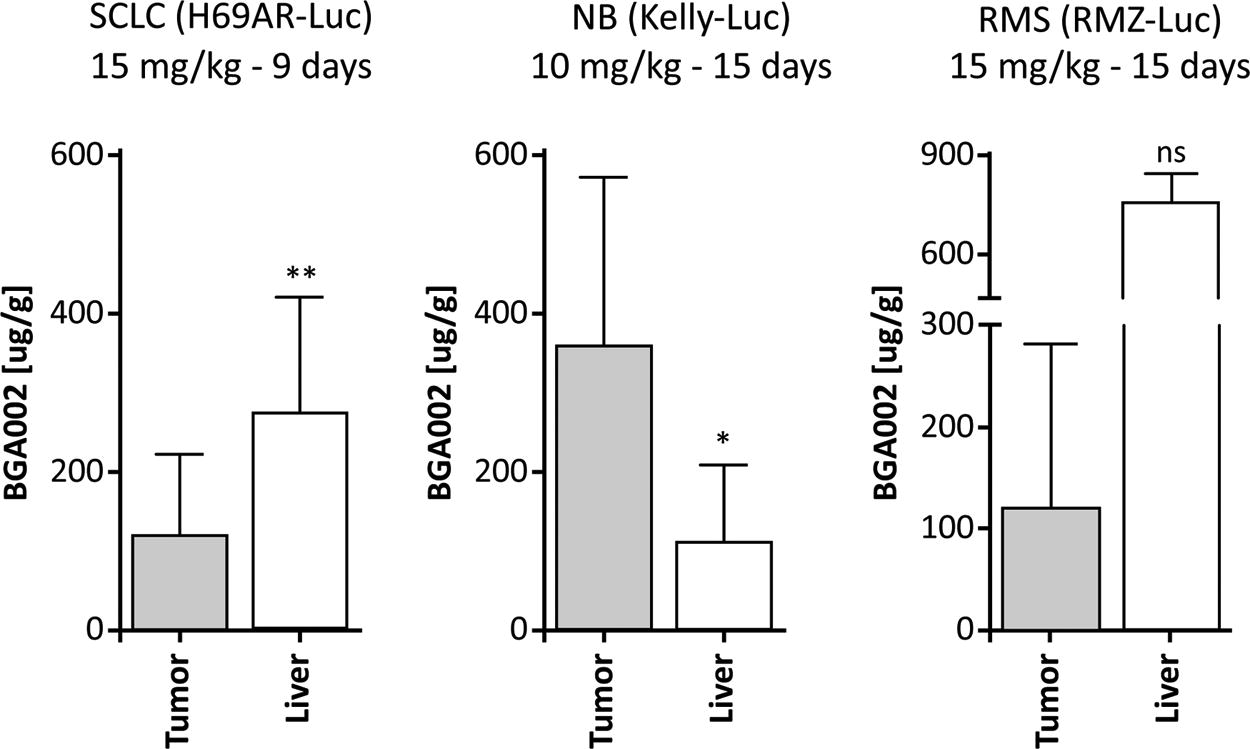
BGA002 is localized in different MNA-positive tumors. BGA concentrations in tumor and liver in MNA-positive SCLC, NB, and RMS mouse models after systemic SC administration of 15 mg/kg BGA002 for 9 days (SCLC), 10 mg/kg for 15 days (NB), and 15 mg/kg for 15 days (RMS). Mean values for SCLC are, respectively, 120 µg/g and 273 µg/g (P value = 0.0072); mean values for NB are, respectively, 360.6 µg/g and 111.9 µg/g (P value = 0.0424); and mean values for RMS are, respectively, 121.4 µg/g and 754.6 µg/g (P value = 0.1). Mann–Whitney test was performed for statistical analysis (ns = P value > 0.05; * = P value ≤ 0.05; ** = P value ≤ 0.01). MNA, MYCN amplification; NB, neuroblastoma; RMS, rhabdomyosarcoma; SCLC, small-cell lung cancer.
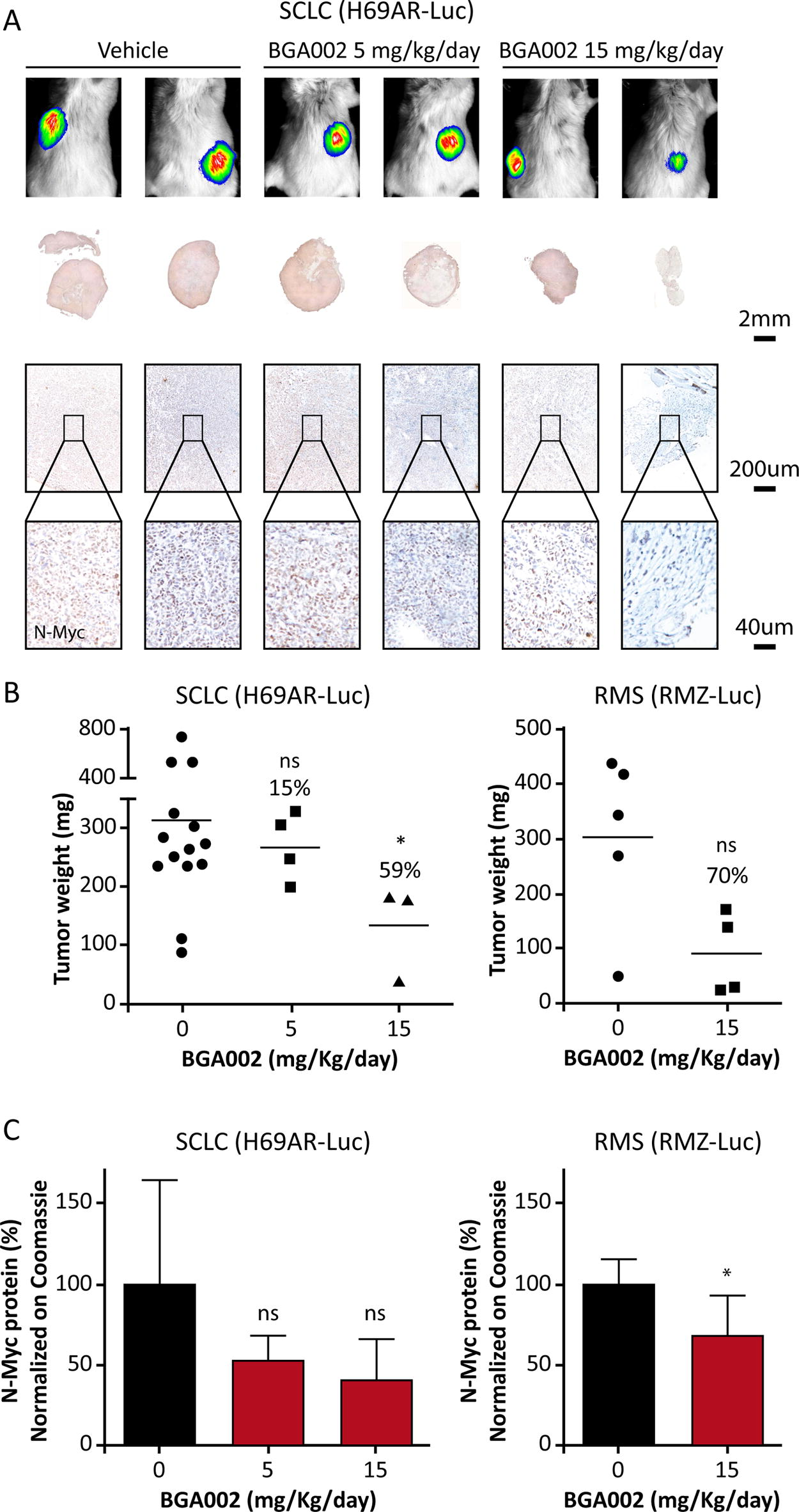
BGA002 induces reduction in tumor weight and N-Myc protein in MNA-positive tumor mouse models.
Discussion
Here we report for the first time the development of a novel specific hybridization-based ELISA approach for the characterization of the PK profile of the MYCN-inhibitor BGA002 and its quantification in biofluids and in the organs of healthy mice and tumor xenografts.
PK analysis in plasma after single administration of BGA002 in mice showed dose-independent, apparently linear PK across the dose range tested, with no evidence of sex differences between male and female animals. Results also show good bioavailability, as minor differences were observed between IV and SC route of administration. The similarity in the concentration range achieved after IV and SC provides evidence that SC administration does not contribute to interindividual variability in exposure. It also supports the use of SC route for further evaluation of efficacy, overcoming potential technical problems of daily IV administrations through tail vein in mice. Moreover, the relatively high bioavailability of BGA002 following SC administration also opens the possibility to use SC as a route of BGA002 administration in human clinical studies. This would ensure better adherence to treatment and lower the burden of treatment owing to the requirements for IV administration.
The PK tissue biodistribution in mice after single subcutaneous administration of [14C]-Ac-BGA002 showed a broad biodistribution in several tissues and organs, with drug levels detected in the kidney, liver, spleen, bone marrow, lymph nodes, and adrenals. This is in line with previous PK studies, 19 which indicated that PNAs conjugated to short synthetic peptide carriers were rapidly cleared from circulation and distributed to a variety of tissues, with highest concentration in liver, kidney, spleen, mesenteric lymph nodes, and adipose tissue (a very similar set of organs to the one we found for BGA002, except to the adipose tissue, which in our study is not one of the main organs where BGA002 accumulates). Overall, these data show that peptide-conjugated PNAs overcome the common issues associated with unmodified PNAs, including poor biodistribution, and they represent a synthetically feasible approach to improve their physicochemical and biological properties.
The broad biodistribution of BGA002 in several organs and tissues could suggest its potential to reach primary tumor and metastasis in different sites in the body. In this respect, it is worth mentioning that BGA002 was also detected in the adrenal medulla (i.e., a preferential primary site of origin for NB) and in the bone marrow (i.e., a preferential site of metastasis for NB and SCLC). Moreover, the observed biodistribution profile suggests a wider application of the class of PNA oligonucleotides covalently bond to the positively charged NLS peptide, which could be used to treat other tumors and even nononcological diseases affecting these different body sites. In contrast, the low biodistribution of BGA002 in the spinal cord and brain indicates that the blood–brain barrier limits its access to the central nervous system (CNS), most probably because the NLS delivery peptide is positively charged (contain lysine and arginine amino acids). If required, for therapeutic indications requiring CNS biodistribution, naked PNA oligonucleotides for their neutral characteristics can cross the blood–brain barrier and reach the CNS. 41
In the present work, we investigated the PK profiles of BGA002 in different MNA and MYCN-expressing tumors. We selected NB and RMS (as relevant examples for poor prognosis MYCN-positive pediatric tumors), and multidrug-resistant SCLC (among the poor prognosis MYCN-positive adult tumors). This specific choice was addressed by the model characteristics themselves, considering their aggressiveness and pathology affinities. In particular, event-free survival commonly shows an endpoint within 60 days.2,13 Although this value tends to be higher in the SCLC model, where we also found evidence of high vascularization, the NB model usually reaches the endpoint value under 30 days, highlighting how relevant MNA is for poor prognosis. Tumor growth rate behaves in a similar way, reaching early relevant volume (500 mm3 within the first 2 weeks) with an exponential growth.2,13 Positive staining to Ki67 and MYCN protein is also a characteristic normally well retained in our models, allowing for proper evaluation of tumor evolution under treatment.2,4,13
Variability was also high in our experimental protocols, but the pharmacological effects were dose-and exposure-dependent,2,4,13 highlighting the feasibility of this model for further evaluation of PK and PD. In fact, we previously reported the in vivo efficacy of BGA002 in an MNA-NB murine model, 2 where we found that SC administration for 15 days of 2.5, 5, or 10 mg/kg/day resulted in a potent dose-dependent tumor growth inhibition that reached tumor elimination after treatment with 10 mg/kg/day. In this MNA-NB mouse model, treatment with BGA002 at 5 mg/kg/day yielded a consistent reduction of the N-Myc protein (evaluated by IHC) and of the tumor vessels. 2
Remarkably, data on the PK profile of BGA002 in tumor across all three highly aggressive MNA tumor mouse models showed drug presence in significant concentrations, with a remarkable threefold higher concentration in NB tumor compared with liver. Moreover, the concentration of BGA002 in tumors was associated to an antitumor PD activity in all the three MNA tumors. In fact, the highest concentration found in NB was also associated to the highest antitumor activity. 2
In summary, MYCN plays a central role in driving tumor evolution, orchestrating several aggressive oncogenic features. Among them, the role of MYCN in neovascularization42,43 has been established. Therefore, evidence from our experimental protocols showing that BGA002 distributes into MYCN-expressing tumor might be also linked to the MYCN role in promoting the neovascularization (and thus accessibility) of the tumors. In this respect, it is worth emphasizing that treatment of multidrug-resistant SCLC tumors with BGA002 led to elimination of tumor vascularization, similar to our previous findings in NB and SCLC.2,13
From a clinical perspective, it should be highlighted that MNA tumors are characterized by a very high amount of N-Myc protein and high-risk and metastatic conditions. Therefore, the treatment schedule used here was based on daily administration of BGA002. For this reason, we would propose frequent administration also in human clinical trials. BGA002 could be administered as a monotherapy or in combination with selected anticancer drugs. In fact, BGA002 was shown to act in synergy with retinoic acid (RA) in MYCN expressing NB, by overcoming RA resistance and restoring RA efficacy. 14 This may be feasible as evidence from preclinical GLP toxicology studies following repeated administration of BGA002 (4 weeks daily) at a higher exposure range than what was described in the current investigation showed that treatment is well tolerated (data not shown).
There is limited preclinical research on the PK of oligonucleotides in tumors in vivo, and none has evaluated the effect of systemic repeated dosing treatment.44–46 Oligonucleotide therapeutics are emerging as promising anticancer precision medicines, especially for the targeting of the large number of undruggable proteins (such as that codified by MYCN) and noncoding RNAs.47 The current findings indicate that the MYCN-specific antigene PNA-peptide BGA002 highly concentrates in aggressive MYCN-related tumors, resulting in significant antitumor activity. Our data reinforce the potential role of oligonucleotides in cancer therapy and pose the basis for the FTIH study with BGA002 in these highly aggressive MYCN-positive tumors. BGA002 already obtained orphan drug designation for NB by the Food and Drug Administration (FDA) (orphan register: DRU-2017–6085) and the European Medicines Agency (EMA) (orphan drug application: EMA/OD/020/12), for soft-tissue sarcomas (including RMS) by EMA (orphan drug application: EMA/OD/037/16), and for SCLC by FDA (orphan register: DRU-2018-6260).
Footnotes
Acknowledgments
The authors thank Wissem Eljeder for suggestions and comments. They also thank the following contract research organizations for their skilled support: Glp Life Test, Charles River, Selcia, Aptuit, Farefarma, and WilResearch (now Charles River).
Author Disclosure Statement
R. Tonelli is BIOGENERA shareholder. A.L. Scardovi worked for BIOGENERA at the time of the studies; she is currently working for Ritrova Therapeutics, Inc. D. Bartolucci, S. Bortolotti, S. Angelucci, C. Amadesi, G. Nieddu, and L. Cerisoli are working at BIOGENERA. No potential conflicts of interest were disclosed by the other authors.
Funding Information
R. Tonelli and P. Hrelia are funded by the University of Bologna (ECOITONELL.). L. Montemurro is funded by AGEOP OdV. A. Scardovi, D. Bartolucci, S. Bortolotti, S. Angelucci, C. Amadesi, G. Nieddu, and L. Cerisoli were funded by Biogenera SpA at the time of the studies. Sean Oosterholt and Oscar Della Pasqua received no funding for their contribution to this project.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.