
Abstract

Introduction
In 2022
My life's work was carried out in many laboratories, from Paul Zamecnik's laboratory at the Worcester Foundation for Experimental Biology (now part of the University of Massachusetts Chan Medical School) to Hybridon, a company I joined as a founding scientist with Paul in 1990, and finally, Idera Pharmaceuticals. Many colleagues and collaborators have contributed to the advances I made, too many to name all individually, but please find their names in the references cited and know how much I value all their contributions. Research is always a team effort, a fact that is too often forgotten when the senior contributor is recognized.
In 2017, after a long career, I retired from Idera Pharmaceuticals with the intention of taking the slower lane, but still wanting to keep my hands in, I agreed to write some reviews and edit a book. This, in turn, caused me to look back over my old research and I started to reconsider past results with our current knowledge in mind. I realized that we—the field of oligonucleotide therapeutics—had taken one-way streets at certain points in our research and neglected to investigate other perhaps less obvious side roads. Roads, that now, with hindsight and more knowledge, seem much more promising and perhaps will lead the field in new and even more successful directions. Without the pressures and responsibilities of a full-time job, I could investigate and experiment at my own pace, so I founded Arnay Sciences to take a look at these other roads.
However, to explain how and why the field took these particular one-way streets and ignored other roads, we need to go back and understand the relevant research in the context of its time and how this understanding has evolved since then. Join me in exploring the history of oligonucleotide therapeutics from my personal perspective—I will show you how oligonucleotide chemistry has really driven progress in the field and how my and others' research contributed to making oligonucleotide drugs a reality.
A Momentous Phone Call
My journey in oligonucleotide therapeutics really started when I joined Michael Gait's laboratory at the world-renowned Laboratory of Molecular Biology (LMB) in Cambridge, United Kingdom, in 1985. In his laboratory, I was exposed to not just the chemistry of oligonucleotides but also the biology. This being the LMB, the environment was overwhelming with all manner of scientific and collaborative endeavors—there was so much to learn! My focus was on the solid-phase synthesis of oligonucleotides and chemical conjugations. At the time, we used a state-of-the-art four-column Omnifit setup to synthesize oligonucleotides using phosphotriester chemistry [1]. It would take 10 h to synthesize a 20-mer and about a week to perform the protection and purification steps necessary for the required quality.
It was during this period that we introduced amino-functionalized oligonucleotides for easy conjugation with fluorescent dyes and biotin [1]. We used these biotin-labeled oligos for nonradioactive diagnostics and fluorescent-labeled oligonucleotides for sequencing [2,3], but unfortunately, the LMB deemed this of little commercial use and declined to pursue a patent. Nowadays, of course, such amino-functionalized oligonucleotides are routinely and widely used in research, and reagents are readily available commercially.
Daniel Brown was working on the opposite laboratory bench to mine. He had retired from his formidable career in the University of Cambridge chemistry department and was spending some time at the LMB. We often discussed the chemistry of oligonucleotides over lunch or afternoon tea. In June of 1986, he mentioned receiving a call from Paul Zamecnik, who asked if he could recommend a good oligonucleotide chemist. Dan told me that he, Paul, and Gobind Khorana had worked together in the mid-1950s in Lord Alexander Todd's laboratory in Cambridge.
Prof. Khorana, as he became known to me, had been awarded a Nobel prize in Physiology and Medicine in 1968, and was incredibly well known in our native India. His picture was in my undergrad biochemistry textbook, and his success encouraged me to pursue a career in chemistry. Coincidentally, I had met Prof. Khorana the previous year when he asked me for directions to the dining hall at Cambridge University during a symposium in his honor. Mike Gait had also worked as a postdoc with Prof. Khorana for several years—it really was quite a small world in oligonucleotide chemistry then, and so through Mike, Dan, and Paul, I came to know Prof. Khorana as well. Along with Mike, Dan, and Paul, he became a mentor for me and later wrote prefaces for two books I published.
Anyway, Dan told me that he had recommended me to Paul as an oligonucleotide chemist so I should expect a call from him. In the call, Paul talked about his 1978 work on using oligonucleotides to inhibit virus replication [4,5] and his recent exciting work on using the same approach as a potential therapy for HIV-1 [6]. He mentioned the word antisense, which stuck with me. After a brief conversation, he offered me the opportunity to advance this research further in his laboratory. I asked if I could read the HIV article before making a decision, and he agreed to mail me a copy.
While waiting for this, I went to the library to read his two articles from 1978 [4,5]. Two weeks later, Paul called me again and inquired what I thought about the article [6] and if I had any question. He described the next steps in antisense research as he saw them and told me that he would love to see me in his laboratory to perform that work. Paul was very humble about his work and able to explain complex science in simple words, so I was inclined to accept the offer. However, Paul would not allow me to say yes or no on the call, instead he insisted on mailing me an offer letter and giving me time to consider. I did not realize then how transformational this call and the follow-up journey would be for me.
Needless to say, I consulted Mike and Dan about the offer. Both encouraged me to accept, so I joined Paul's laboratory in January of 1987. The day before I arrived, Massachusetts had received two feet of snow, so the surroundings were stunningly beautiful, if unfamiliar to me, having grown up in India and coming from the south of United Kingdom. Over the next few days, as we discussed the science and how we should proceed, Paul stressed that we needed to increase the nuclease stability of the oligonucleotides, while maintaining affinity with the target RNA and aqueous solubility.
Phosphorothioate Oligodeoxynucleotides: First Generation Antisense
As Paul suggested, we focused on modifying the internucleotide linkages to increase the stability of oligonucleotides against nucleolytic attacks from 1987 to 1989. We prioritized three internucleotide linkages—methylphosphonate (PM-DNA), phosphorothioate (PS-DNA; Fig. 1), and phosphoramidates (PN-DNA).

Chemical modifications identified during our research for use in antisense and other RNA therapeutics. Details of modifications compared to PO-DNA are highlighted in red. PO-DNA, phosphodiester DNA.
Paul Tso and Paul Miller had published on PM-DNA analogs of oligonucleotides in the early 1980s [7], but to synthesize sufficient amounts of 15–20-mer PM-DNA oligonucleotides on the newly available automated synthesizers, we first had to develop methylphosphonamidite chemistry [8]. PS-DNA analogs and their enzymatic characteristics had been published by Fritz Eckstein and colleagues in the 1970s [9], but again, we first had to establish how to synthesize PS-DNA and PN-DNA analogs on the automatic synthesizers.
To evaluate the characteristics of PM, PS, and PN DNA backbones, we synthesized 15–20-mer antisense sequences complementary to HIV-1. We prioritized antisense sequences based on a study of phosphodiester DNA (PO-DNA) targeted at various regions of HIV-1 [10]. We assessed the sequences for nuclease stability, affinity with the target sequence, and solubility [11,12]. Synthesis of PM and PN backbones was complicated by poor aqueous solubility, but both were more resistant to exonucleases than PS, which in turn was more stable than PO. Affinity to the target RNA, as assessed by melting temperature, showed the opposite: PO had the highest affinity, followed by PS, PM, and PN, which exhibited severely reduced affinity.
These studies demonstrated that none of the analogs had all the desired characteristics. Our next step was to evaluate the PS, PM, and PN antisense oligonucleotides for inhibitory potency in HIV-1-infected cells, in collaboration with Robert Gallo's laboratory at the NIH. All three analogs showed increased potency compared to PO-DNA oligonucleotides, but PS-DNA was certainly the standout [11,12]. The results demonstrated that the increased nuclease stability of PM-DNA and PN-DNA did not directly translate into increased potency, suggesting that nuclease stability was not the only factor in antisense activity. We reasoned that perhaps affinity and aqueous solubility also had an impact.
So our focus shifted to investigating the PS-DNA modification in more detail. We synthesized several antisense sequences complementary to HIV-1 as well as control sequences. Curiously, inhibition of HIV-1 replication in HIV-infected cells was more profound in the antisense-treated cells, but was also seen with control sequences [13,14]. We hypothesized that the polyanionic nature of PS-DNA might result in unspecific binding to the cell surface and thus interfere with the binding and entry of HIV-1. Julianna Lisziewicz from Gallo's laboratory established chronically infected cells that avoided the HIV-1 binding and entry steps to address this concern. In this assay, PS-DNA antisense showed largely sequence-specific anti-HIV activity and thus established that PS-DNA was a viable chemical modification for antisense oligonucleotides [15,16]. We obtained similar results with antisense PS-DNA for the inhibition of influenza virus replication [17].
However, it was still unclear why PS-DNA antisense showed so much more potent antisense activity than PM-DNA or PN-DNA. In collaboration with Sandra Mayrand in Thoru Pederson's laboratory, we targeted a U1-RNA as well as an exogenously added 514-nucleotide RNA in HeLa cell nuclear extracts with our modified antisense oligonucleotides. Only the PS-DNA antisense oligonucleotides formed RNA-DNA duplexes with the target RNAs that could recruit endogenous RNA-DNA duplex-activated RNase H and were cut by the enzyme [18]. However, the efficiency of this process remained lower for PS-DNA than PO-DNA. When we combined PS-DNA segments with PM-DNA or PN-DNA segments within the same antisense oligonucleotide, RNase H-mediated cuts only happened within the PS-DNA segments. We referred to this cutting as “restriction endonuclease-like cleavage.”
Notably, this was the first time anyone had combined segments of different chemical backbone modifications within PS-DNA—keep in mind that at this point in time (late 1980s), making these oligonucleotides was still challenging and time-consuming. Automatic oligonucleotide synthesizers had only just become commercially available.
Our experiments demonstrated that it was possible to combine the desirable characteristics of different modifications within the same oligonucleotide—we named them “mixed-backbone oligonucleotides” (MBOs). However, the antisense activity of these MBOs was not encouraging, and we, therefore, did not pursue this side road any further [19].
In 1989, we also studied the in vivo pharmacokinetic profiles of S35-labeled PS- and PM-DNA after intraperitoneal administration in mice. The PS-DNA demonstrated a very short plasma half-life and was eliminated through urinary excretion over 3 days. PM-DNA was also eliminated through urinary excretion within a few hours, not days. Considering the results from the HIV infection experiments, these findings suggested to us that the unspecific binding of PS-DNA to proteins might delay elimination, and PM-DNA was eliminated due to poor binding to proteins.
Still, the very short plasma half-life was concerning since it meant we would have to improve the administration route or increase the dose to maintain the plasma concentration necessary for effective therapy. We also wondered where the PS-DNA went in between being eliminated from the plasma compartment and excreted from the body. So we monitored plasma clearance by sampling blood, organs, urine, and feces at multiple postdosing times, from 5 min to 48 h [20]. Both intraperitoneal and intravenous administration resulted in a plasma half-life of about 1 h. Still, PS-DNA was widely disposed to various organs, and elimination from these organs was slow. However, the pharmacokinetics were calculated based on S35 levels and do not reflect the integrity of the PS-DNA. Polyacrylamide gel (PAGE) analysis of various tissues demonstrated the presence of intact PS-DNA and a ladder of degradation products primarily caused by exonucleases and not endonucleases [20,21].
To determine if the degradation was caused by 3′ or 5′ exonucleases, we studied the pharmacokinetics of 3′- and 5′-capped PS-DNA [22]. These PS-DNAs demonstrated similar plasma half-life, tissue disposition, and elimination in urine, but PAGE analysis showed that the 3′-capped PS-DNA was significantly more stable. Clearly, primary degradation happened at the 3′ end. We now know that the pharmacokinetics of PS-DNA sequences remain similar across rats [23], monkeys [24], and humans [25], and are largely independent of their sequence, except in cases where protein binding is affected [26]. For example, plasma clearance and tissue disposition of PS-DNA are impacted by GGGG motifs [21,27] due to quartet formation or pre-dosing with aspirin [28], which reduces protein binding.
Our next step was to conduct safety studies in animal models. In mice and rats, subcutaneous administration of PS-DNA was well tolerated, but resulted in dose-dependent changes in clinical chemistry and caused gross necropsy [29–31]. Histological analysis of selected organs showed the presence of inflammatory cells. In primates, bolus intravenous administration of even low doses of PS-DNA (2.5 mg/kg) led to severe hemodynamic changes.
Due to these unexpected observations, we paused the clinical development and conducted more studies in dogs and pigs. In these animals, treatment with PS-DNA resulted in minimal or no hemodynamic change, suggesting that the hemodynamic changes were specific to primates (Agrawal, unpublished data). We conducted extensive work with different PS-DNA lengths, sequences, and varying infusion rates. We could only conclude that PS-DNA activated complement, prolonged partial thromboplastin time, and caused thrombocytopenia and leukopenia [32]. At the time (the early 1990s), assays to evaluate complement cascades were limited to specific laboratories; Patricia Giclas kindly analyzed the samples for us.
The safety signals were dependent on the dose and rate of infusion, and slow infusion of PS-DNA avoided the hemodynamic changes. When we reported these findings in 1994 [32], the FDA published guidelines on the administration of PS-DNA in humans and included primates as one of the species required for IND-enabling studies [33]. The guidelines recommended that bolus IV administration of PS-DNA should be avoided.
At this stage (1993), our development candidate was a PS-DNA 25-mer antisense targeted to the gag-region of HIV-1, called GEM91 [34–39]. To manufacture the required quantities under good manufacturing practices (GMP) conditions, we set up the first commercial oligonucleotide manufacturing unit, Hybridon Speciality Products (HSP), in Milford, MA. In 2000, HSP was sold to Avecia, which now operates under the name of Avecia Nitto Denko at the same location [40].
In 1994, we initiated the first pharmacokinetic study of PS-DNA in humans [25]. We treated HIV-infected volunteers with S35-labeled GEM91 and paid particular attention to potential complement activation. As in the primates, plasma half-life was under an hour, and urinary excretion was the primary route of elimination. When we administered GEM91 as a 2-h IV infusion at doses of 0.1–1.0 mg/kg, treatment was well tolerated. With subcutaneous administration, 33% of volunteers developed localized painful nodules at the site of injection, which resolved without intervention [41].
We also investigated if repeat dosing could increase retention with 2-h IV infusions every other day for 27 days (14 × 0.5–2.0 mg/kg), but there was no change. To give us the best chance of seeing anti-HIV activity, we administered GEM91 by continuous IV infusion over eight days (0.2–4.6 mg/[kg·day]), maximizing the dose, while minimizing complement activation. Treated volunteers developed dose-dependent injection site reactions, including induration, flu-like symptoms, and thrombocytopenia. When we analyzed HIV levels in collected blood samples, levels remained unchanged in the control group, but there was a dose-dependent increase in HIV levels in the treatment group from days 4–6 [36]. Although this returned to baseline by day 8, we stopped dosing immediately!
We were totally baffled by this result and considered various theories. Our first hypothesis was contamination in the drug, but thorough analysis showed no contamination or endotoxin. We reluctantly concluded that the cause was either the specific GEM91 sequence or that it was a class effect of PS-DNAs. With over 20 PS-DNA antisense candidates in clinical development, the whole field really needed to know which one it was urgently! The suspicion that it was a class effect caused fierce discussion among scientists and predictions that the field was doomed among stock analysts. This made the years from 1996 and afterward extremely difficult for the field as raising further capital for continuing research became very difficult.
To determine if the problem was specific to the GEM91 sequence, we started to truncate GEM91 from the 5′ end. With these experiments, we were able to demonstrate that the TCG triplet close to the 5′ end (5′-CTC
With the increasing amount of data on PS-DNA antisense, one question kept coming up: What is the difference between the Rp and Sp stereoisomers of PS-DNA? Due to the chiral nature of PS-DNA, each PS linkage in a given molecule may be either of these isomers. Any particular linkage may contain the same or a different stereoisomer in another molecule of the same sequence such that all PS-DNA products contain a mixture of distinct stereoisomers (2n−1 maximum possible; n = nucleotides).
We first synthesized stereo pure Rp oligonucleotides using an enzymatic approach [43], but to obtain larger quantities, we developed nucleoside bicyclic oxazaphospholidines as novel synthons [44]. Each isomer showed different characteristics, for example, affinity to target RNA Rp>Sp, nuclease stability Sp>Rp, and RNase H activation Rp>Sp. We also studied stereo pure PS-DNAs with defined isomers placed at specific positions, but activity did not improve enough to justify the additional production cost [45,46]. Of note, the Sp isomer demonstrated increased protein binding, resulting in complement activation at very low doses.
Over the next years (1996 onward), clinical development of many PS-DNA antisense was discontinued since most of these sequences contained CpGs [47]. The mechanism of action was in question—was the efficacy seen in viral and cancer models genuinely antisense or simply immune activation, or a bit of both? Was there even such a thing as RNase H antisense? To illustrate, our articles' titles from that time were “Antisense Therapeutics: Is it as Simple as Complementary Base Recognition?” [48] and “Importance of Nucleotide Sequence and Chemical Modification of Antisense Oligonucleotides” [49]. Funding from Wallstreet and pharma partners dried up and consequently, advances were slow.
Phosphorothioate Oligoribonucleotides and 2′-Substituted RNA: Splice Modulators
In 1989, parallel to pursuing PS-DNA antisense, we also studied PS oligoribonucleotides with 2′-substitutions (PS-2′RNA; Fig. 1). These analogs showed higher affinity to complementary RNA as well as increased nuclease stability compared to PS-ODNs, but failed to activate RNase H. They were less potent in our HIV-1 antisense experiments [50,51], but to us, this was simply further confirmation that RNase H activation was essential for target RNA knockdown. Based on these results, we initially did not go further along this road, but we later realized that phosphorothioate oligoribonucleotides (PS-RNA) and PS-2′RNA could be used to modulate RNA processing by simply blocking access to the target sequence. For example, we used PM-DNA, a non-RNase H substrate, to mask U1 and U2 snRNPs required for spliceosome assembly [52].
At a conference in the mid-1990s, Ryszard Kole mentioned his recent article where he had used phosphodiester 2′-O-methyl oligoribonucleotide antisense to modulate splicing in cell-free systems. We decided to collaborate and evaluate PS-2′RNA for its ability to modulate splicing in intact cells. As stated in the introduction of the article we published together in 1996 [53], “It was not at all clear whether the oligonucleotide delivered into the cell could enter the nucleus, hybridize to the aberrant splice site in competition with the splicing factors, and promote the formation of the spliceosome and subsequent splicing at the correct splice site.” The article demonstrated that all this did in fact happen and thus was the first example of modulating splicing with antisense, establishing a totally new antisense mechanism.
Following the publication, Steve Wilton, who was working on Duchenne Muscular Dystrophy (DMD), approached us to collaborate on modulating the splicing of the DMD gene. These studies were very successful and as we wrote in the resulting article [54], “AOs (antisense oligonucleotides) may be regarded as a highly specific nucleic acid drug that re-programs existing cellular machinery to by-pass or minimize the consequences of serious dystrophin gene mutations. This antisense therapy has the potential to reduce the severity of the disease so that a boy with a DMD genotype would only develop a milder Becker syndrome.” It gives me great pleasure that several splice modulating antisense drugs resulting from this research have been approved for the treatment of DMD in the last 10 years.
During these studies, we also found that PS-RNA and PS-2′RNA had a much more favorable safety profile than PS-ODNs [30,51,55–59]. So, we wondered—how could we use these very desirable characteristics in RNase H-based antisense, despite PS-2′RNA forming RNA/RNA hybrids that RNase H could not recognize?
Hybrid/Mixed Backbone/Gapmer Antisense: Second/Third-Generation Antisense
Based on our prior experience of using mixed PS-DNA, PM-DNA, and PN-DNA segments in an oligonucleotide, we thought that perhaps we could combine the desirable characteristics of PS-DNA and PS-2′RNA in a similar way. We explored various configurations, such as placing a segment of PS-2′RNA on the 3′ end, on the 5′ end, or both the 3′ and 5′ ends, while retaining PS-DNA linkages in the other parts [30,51,55,56,59]. We knew from our previous studies that a stretch of 8 PS-DNA bound to the target RNA was needed to optimally activate RNase H [59].
We had also established that in vivo degradation primarily occurred at the 3′ end [22], so we knew we would increase nuclease stability by placing segments of PS-2′RNA on at least the 3′ end, if not both. Based on the available data, we focused exclusively on the design with PS-2′RNA segments on both the 3′ and 5′ ends, with a PS-DNA segment in between. We called this new type of antisense oligonucleotide “hybrid” to differentiate them from our previous mixed-backbone sequences [51]. They are now more commonly called gapmers (Fig. 2).
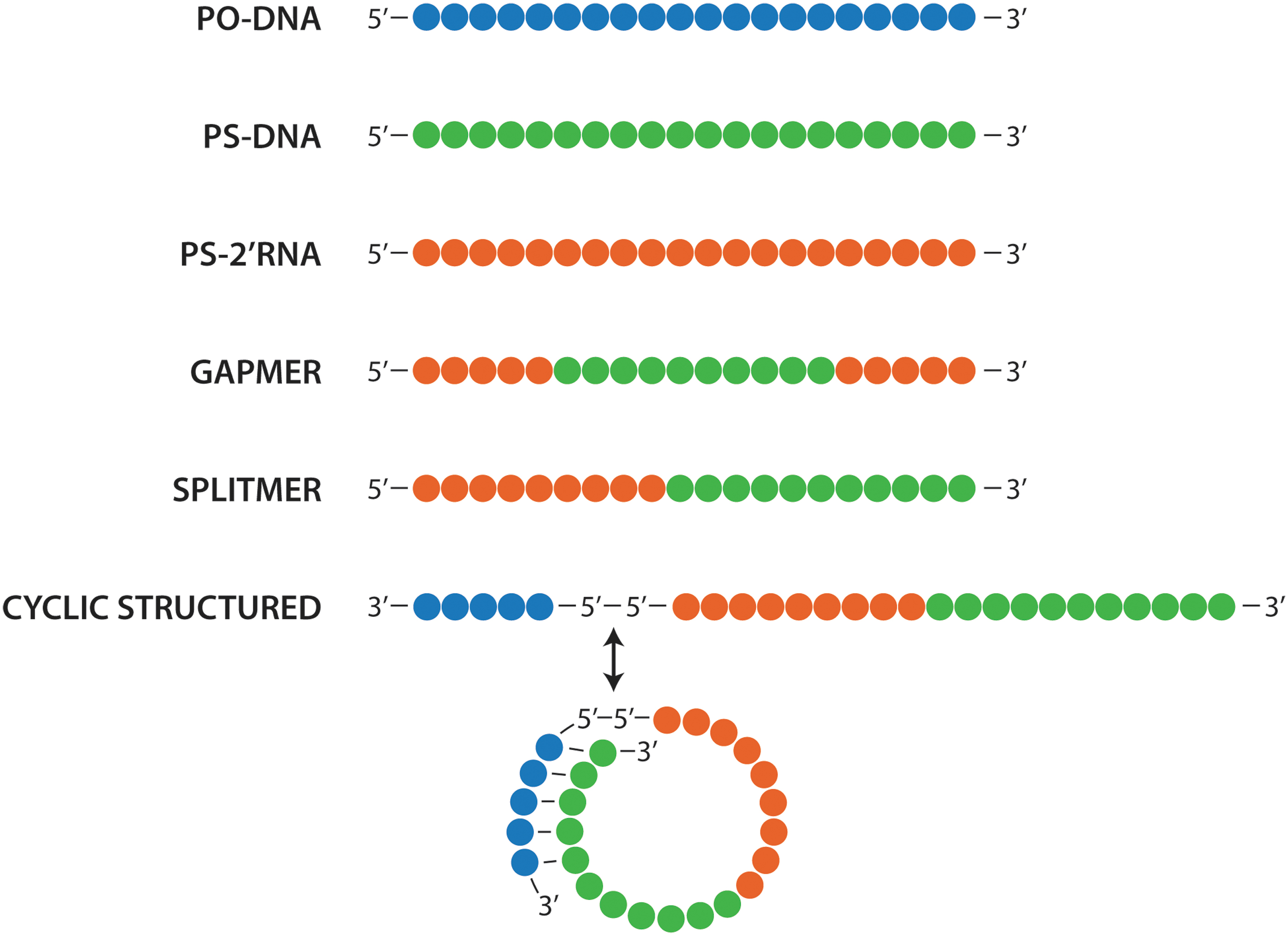
Antisense designs discussed here. Composed of PO-DNA (blue), PS-DNA (green), and PS-2′RNA (red). In gapmer antisense, PS-DNA is placed in the center flanked by PS-2′RNA on both 3′ and 5′ ends. In splitmer antisense, PS-DNA is placed on the 3′ end of the antisense and PS-2′RNA on the 5′ end. In cyclic structured antisense, splitmer antisense is linked to a circularizing domain by 5′-5′ linkage. The circularization domain could comprise PO-DNA or PS-DNA, or a combination thereof. PS-DNA, phosphorothioate oligodeoxynucleotides.
During the early 1990s, our focus was very much on the clinical development of GEM91 and work on gapmer antisense was put on the backburner. However, in 1992, we filed a patent application on our new gapmer antisense designs, which was granted and resulted in several US patents (5,652,355; 6,143,881; and 7,045,609) and their worldwide counterparts [60]. These patents covered gapmer antisense designs comprising PS-DNA segments in combination with 2′-substituted RNA or DNA segments, including, but not limited to 2′-O-methyl, 2′-O-methoxyethoxy, and 2′-alkoxyalkyl substitutions, as well as gapmers with phosphodiester and phosphorothioate linkages within the 2′-RNA segments [61].
With the discontinuation of GEM91 in 1996 due to unexpected immune activation and safety concerns, our focus shifted to exploring these gapmer designs. Extensive characterization studies demonstrated significant advantages of these second-generation designs in multiple gene and disease targets. Notably, they showed increased potency and in vivo stability, as well as reduced inflammatory and polyanionic responses [29,30,32,51,55–58,62,63]. We even pursued oral and rectal delivery based on the massively increased in vivo stability [64,65]. To replace GEM91, we developed the gapmer GEM92 for HIV and additionally developed GEM231 for cancer therapy.
The development of the gapmer antisense chemistry breathed new life into the field, and an increasing number of articles successfully using these designs started to appear. In 2001, ISIS Pharmaceuticals, now renamed to IONIS, obtained a co-exclusive license for the gapmer antisense chemistry and other related patents from us (Hybridon) [66].
Currently, gapmer designs with segments of PS-DNA and 2′-O-methoxyethoxy for RNase H-mediated knockdown are widely used and have enabled several successful drug approvals [67,68].
The Discovery of Pattern Recognition Receptors: Nonspecific Receptor-Mediated Immune Activation
As already discussed, we had determined that the TCG sequence in GEM91 induced a sequence-specific immune response in primates and humans that resulted in systemic responses, including an increase in HIV viral load [36,69–71]. Also, PS-DNA antisense targeting human papilloma virus showed broad-spectrum antiviral activity in animal models, but this was abolished in immunocompromised mice. Our findings were congruent with a few published reports on the immunostimulatory properties of nucleic acids, especially bacterial DNA, but these had been limited to cell cultures and mice [72–75].
Later studies by Krieg and Klinman provided more insights into the immune reaction we observed with the TCG sequence in humans [76]. They showed that unmethylated CpGs in bacterial DNA were associated with immune stimulation, and the subsequent discovery of various pattern recognition receptors (PRRs) during the late 90's and early 2000's explained how this worked [77].
PRRs are a family of proteins expressed on various immune cells that recognize the pathogen-associated molecular patterns (PAMPs) of bacteria and viruses and initiate innate immune responses to protect the host. To date, Toll-like receptors (TLRs), Rig-like receptors (RIG-1), inflammasomes, and STING have been identified as PRRs that recognize nucleic acid PAMPs. The most relevant for oligonucleotide-based therapeutics are TLR3 (for double-stranded RNA), TLR7 (for single-stranded RNA), TLR8 (for single-stranded RNA containing modified bases), and TLR9 (for DNA containing unmethylated CpG-dinucleotides).
In addition, inflammasomes (for double-stranded DNA), RIG-1 (for RNA containing 5′ phosphate), and STING (viral DNA and synthetic dinucleotides) can also impact the outcome of oligonucleotide therapeutics. Of note, the expression profile of PRR is species specific, with different PRRs expressed in different cell types in rodents versus primates and humans. Some of these receptors are expressed on the cell surface and/or in endosomes (TLR3, 7, 8, and 9) and some purely in cytosolic compartments (RIG-1, inflammasome, and STING). These PRRs recognize sequence motifs, secondary structure, or degradation products of nucleic acids, and activate distinct immune cascades depending on the combination of ligand and receptor.
Initially, we really only wanted to identify problematic sequence motifs and optimal positions of additional chemical modifications to avoid such immune activation in our antisense candidates. So we investigated the structure-activity relationships (SAR) of a few representative PS-DNA sequences containing CpGs [78–83]. Compared to controls without, PS-DNA sequences with CpG motifs caused TLR 9-mediated immune responses and cytokine expression, particularly IL-12 and IL-6.
As part of these studies, we acquired a lot of information peripheral to our original intent, but useful. For example, we found that modifications such as 2′-substituted nucleosides, internucleotide linkages, or heterocyclic bases placed immediately upstream of any CpG significantly reduced immune stimulation compared to downstream [84–86]. Such modifications in the N-1 and N-2 positions (upstream or downstream from the CpG) resulted in a complete loss of immune stimulation [78,80,81].
In PS-DNAs containing 2 CpGs placed 10 nucleotides apart, substituting the C of the 5′ CpG with a 2′-O-methyl ribonucleoside mitigated the immune stimulation, despite the CpG toward the 3′ end, while substitution in the 3′ CpG, but not the 5′ CpG, had no effect [87]. Immune stimulation was also mitigated when accessibility to the 5′ end was blocked by linking two PS-DNA using a 5′-5′ linkage, but increased when PS-DNA had two 5′ ends (created by linking two PS-DNAs through 3′-3′ linkage; Fig. 1) [88,89]. Clearly, recognition of CpG motifs in DNA starts from the 5′ end, and blocking accessibility to the 5′ end is critical in avoiding immune activation [90].
We also evaluated a large number of other synthetic nucleosides for CpG substitution [82,91–94]. Most mitigated the immune stimulatory activity of CpG-containing PS-DNA, but a few had no effect. These results demonstrated that the 2-keto, 3-imino, and 4-amino groups of the cytosine, as well as the 1-imino, 2-amino, and 6-keto groups of the guanine in a CpG-motif, are essential for the immunostimulatory activity, while the absence of N7 on guanine has no significant effect (Fig. 1) [82,91–93]. One of these substitutions, 5-methyl-cytosine, is now widely used to modify all cytosines in antisense candidates. Notably, 5-methyl-cytosine substitution of C in CpG not only mitigates immune activation but also converts PS-DNA to act as an antagonist of TLR9 [94]. Other modified nucleosides, including 7-deazaG, araG, and araC, could be used as substitutions for C or G in CpG motifs to maintain immune activation (91–93; Fig. 1).
We also performed similar studies with PS-RNA and PS-2′-RNA to elucidate how these analogs interact with PRRs and influence immune responses. The PS-RNA and PS-2′-RNA engaged TLR7, TLR8, and TLR3, depending on the nucleotide sequence, sequence composition, modified nucleosides, intermolecular and intramolecular structures, and accessibility of the 5′ end [95–98]. Importantly, 3′-3′-linked PS-RNA also showed increased immune activation, and activity was mitigated in 5′-5-linked PS-RNA.
To summarize our findings, a number of modifications can be made to PS-DNA and gapmer antisense containing CpGs to avoid immune activation. These include substituting CpG with synthetic nucleotides, blocking 5′-accessibility, substituting C with 5-methyl cytosine, placing CpG toward the 3′ end rather than the 5′ end, and modifying upstream positions [91–93,99]. These insights have helped the field design the third generation of PS-DNA and gapmer antisense.
At a Crossroad: Oligonucleotide-Based Immunotherapy
As already mentioned, our initial objective was to understand how to mitigate immune activation by our antisense candidates, but we soon realized that what we really had learned was how to create optimal agonists and antagonists for endosomal TLRs. We thought that these might be useful in immunotherapy of cancer, to treat viral infections, asthma, and allergies, or as vaccine adjuvants. Consequently, we designed a library of TLR agonists and characterized the resulting immune responses in mice, primates, and humans [100]. These TLR agonist candidates had two exposed 5′ ends since they were 3′-3′ linked and contained synthetic CpR motifs in which G was substituted with 7-deazaguanosine and ara-G (Fig. 1). Surprisingly, treatment with these agonists triggered a unique immune activation profile that remained similar across all tested species unlike that of CpG motifs [99,101,102].
In healthy human volunteers, one of our candidates, IMO-2055 [103,104], produced immune activation even at the very low dose of 0.005 mg/kg when administered subcutaneously. Weekly (n = 52) subcutaneous injections of IMO-2055 induced an immune response after each injection, suggesting that repeat dosing did not result in the development of tolerance [105]. Notably, there was a cooldown period of 3–5 days after administration, during which repeat administration did not result in immune activation. Treatment with both IMO-2055 and another candidate, IMO-2155, (at doses of 0.2–0.32 mg/kg each) led to a bell-shaped dose-response with induction of suppressive signaling, including expression of IDO-1, at higher doses [106].
Both the cooldown period and the suppression feedback loop make sense since TLR9 activation acts as a danger signal and triggers cytokine expression, which leads to the margination of immune cells from the circulation to the periphery, leaving the circulation depleted of activatable cells. Overstimulation of the immune reaction would be detrimental and thus regulation is necessary. We concluded that dose as well as frequency of dosing were critical for immune activation through TLR9. The implications of our findings for the mechanism of action and safety of antisense therapeutics are clear: typical doses used in antisense therapeutics far exceed the activation threshold of TLR9 and therefore will cause immune activation. It is imperative to avoid such activation and therefore to remove any immune stimulatory motif as much as possible.
We also created oligonucleotide-based antagonists for TLR7 and TLR9 (IMO-3100) and TLR7, TLR8, and TLR9 (IMO-8400 and IMO-9200) [94,107,108]. We observed the therapeutic potential of these antagonists in preclinical models of psoriasis [109], lupus [110], DMD [111], colitis [112], and genetically defined lymphomas [113] and clinical benefit in subjects with psoriasis [114] and Waldenstrom macroglobulinemia [115].
A Few Critical Observations
The SAR studies with both PS-DNA and PS-RNA showed that 3′-3′ conjugation led to increased immune activation, whereas 5′-5′ conjugation mitigated immune activation [88,89,95]. However, what impact does such conjugation have on antisense potency? To answer this, we compared the activity of 5′-5′-, 3′-3′-, and 3′-5′-linked versions with the unlinked parental 19-mer antisense [116]. We expected the 3′-3′ antisense to be more potent based on the increased nuclease resistance we saw in previous experiments. Surprisingly, although, the 5′-5′ linked antisense showed significantly higher activity in both cell culture and mice compared to the other three and even a gapmer control (Agrawal, unpublished data). Yet, all four versions demonstrated similar affinity to the target RNA, PCSK9. How was this possible?
An explanation came from studying the fragments generated by RNase H cleavage of the antisense DNA/target RNA hybrids. The 3′-3′- and 3′-5′-linked as well as the unlinked version generated cleavage products starting from position 8 (counted from the 3′ end of the antisense) all the way to the 5′ end of the bound antisense. Conversely, with the 5′-5′ linked, antisense cleavage occurred exclusively at positions 8–11 of the targeted RNA. If those positions contained mismatches, cleavage was significantly reduced. This little detail later allowed us to selectively target single nucleotide polymorphisms. We referred to these 5′-5′ linked antisense PS-DNAs as gene silencing oligonucleotides, but further characterization showed that the increased length also meant increased adverse polyanionic characteristics, again leading us to abandon a possible sideroad in favor of the more widely accepted hybrid/gapmer design.
Connecting the Dots: Designing the Next Generation of Oligonucleotides
As previously mentioned, in 2017, I retired from Idera Pharmaceutical, a company I co-founded. My former postdoc advisor, Mike Gait (also retired), and I collaborated in editing a book, “Advances in Nucleic Acid Therapeutics” [117]. We also wrote a couple of review articles on the history of antisense [118], and I jotted down the progress in oligonucleotide chemistry made in our and others' laboratories [119]. While going back through my old research, I realized that our gapmer antisense designs and PS-2′RNA modifications were now 25 years old and still being used in approved drugs and clinical development of antisense candidates. Yet these designs and modifications were developed before we understood how synthetic oligonucleotides interact with PRRs! It made me realize that we have yet to connect the insights we gained from our work on antisense chemistry with those from our work on SAR of oligonucleotides and PRRs (Fig. 3).
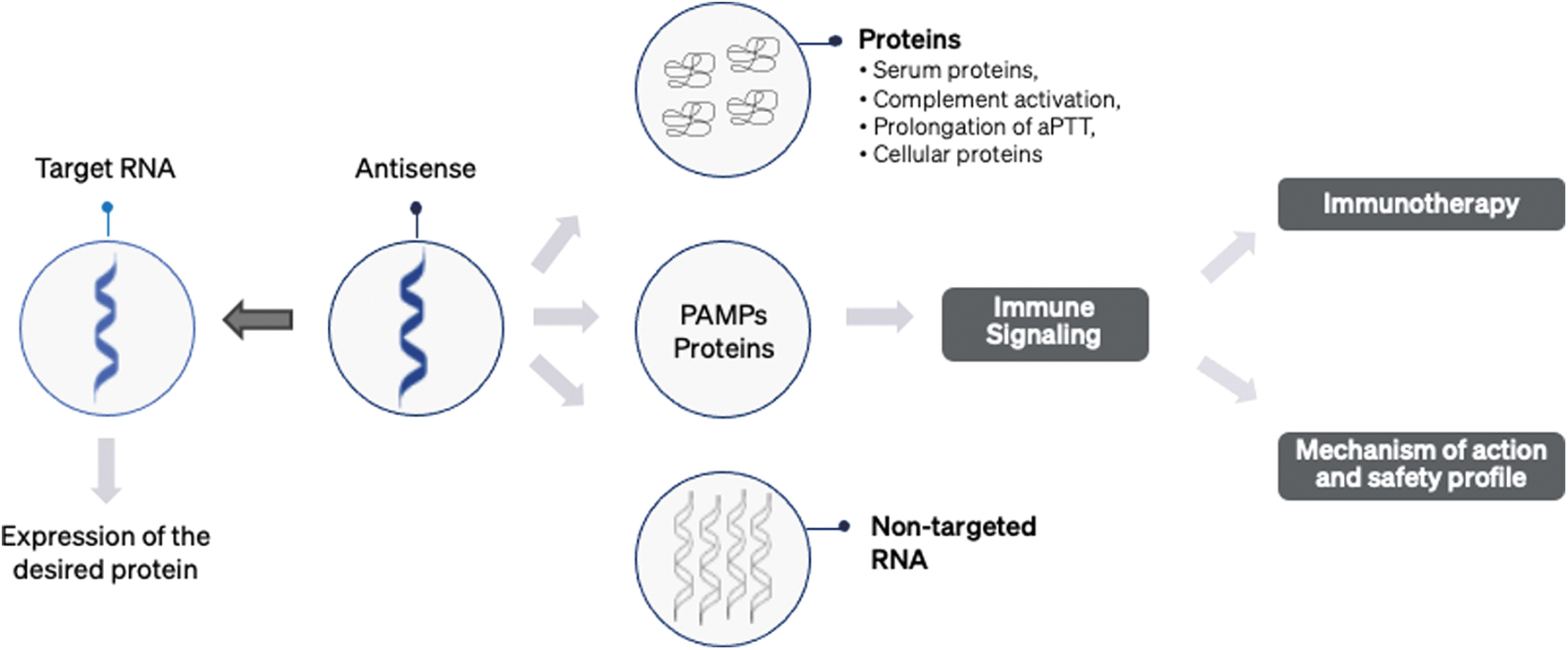
Antisense is designed to hybridize targeted RNA, thereby modulating protein expression, shown on the left side of the antisense. However, antisense candidates have been shown to bind to proteins, PRRs, and nontargeted RNAs, which leads to off-target activity, as shown on the right-hand side of the figure. More importantly, this unspecific binding to targets other than the intended RNA reduces the effective dose of antisense oligonucleotides. PRR, pattern recognition receptor.
When we started exploring antisense in the late eighties, our most important criteria were nuclease stability, affinity with target RNA, and aqueous solubility [119]. Soon we learned that RNase H activation and protein binding are also critical. This led us to identify essential modifications, including PS-DNA (Rp and Sp isomer), PS-RNA, and PS-2′RNA (2′-O-methyl and 2′-methoxythoxy; Fig. 1) and other useful information about modified nucleosides, substitutions of C and G in CpG and 3′-3′ and 5′-5′ linkages (Fig. 1). These findings not only facilitated the development of antisense but also other RNA therapeutics, such as siRNA.
As discussed above, we learned that accessibility of the 5′ end is critical for immune activation and oligonucleotides containing two 5′ ends or two 3′ ends have contrasting characteristics. Oligonucleotide sequence is crucial as it can be recognized as a PAMP, but specific nucleoside modifications of CG, for example, 5-methylC or 7-deazaG (Fig. 1), can mitigate (or enhance) the resulting immune profile. At the same time, I was contemplating the recent progress in targeted delivery with GalNAc, antibody, and fatty acid conjugates and how this advanced the field tremendously. Then I realized that, by using these conjugates for targeted delivery, we could now afford to use chemical modifications or structures that reduce protein binding without losing efficacy. So sideroads that we stopped exploring previously were now looking much more promising. Would the designs we discarded work better than gapmers with targeted delivery? Let me show you what I mean…
Splitmer Antisense
As mentioned in the hybrid/gapmer section, we had briefly explored various configurations with segments of PS-2′RNA on either the 3′ end or the 5′ end, in addition to the standard gapmer design with PS-2′RNA segments on both ends. However, with limited funding and time, we focused exclusively on the gapmer design. With new insights, I thought discarding these other configurations was a mistake as we lacked the necessary details at that time. I reasoned that a “splitmer” design (Fig. 2), consisting of PS-DNA for the first 9–12 nucleotides from the 3′ end and PS-2′RNA otherwise, might have increased activity as well as reduced off-target effects compared to a gapmer [71,120,121].
During our research into the immune response to nucleic acids, we had learned that 2′RNA modifications were needed only on the 5′ end of the oligonucleotide to mitigate interaction with PRRs, and that these could be either on a PS or PO backbone. We also knew that nuclease degradation happened primarily at the 3′ end and that nuclease stability was somewhat reduced if the 3′ end contained PS linkages, but not 2′RNA modifications. However, I thought that this might not be a big disadvantage as slow degradation might avoid tissue buildup after repeat administration. More importantly, exonucleolytic loss of 2–3 nucleotides from the 3′ end would bring the length of the PS-DNA segment available for RNase H recognition below the necessary number and thus reduce off-target activity. Splitmer antisense showed improved potency and specificity than gapmer antisense [120] (Fig. 5).
Cyclic Structure Antisense
We had also previously observed that 5′-5′-linked PS-ODN antisense showed increased potency and specificity, as well as reduced immune responses, but had stopped development due to the increase in adverse polyanionic characteristics that resulted from doubling the length. However, this would no longer be a problem with the targeted delivery options available now. What did remain a potential issue was the accessibility of the exposed 3′ end to nucleases, so I pondered ways of blocking this end in the 5′-5′-linked constructs. Design guidelines for antisense generally recommend avoiding sequences that form intermolecular or intramolecular self-hybridization of more than four bases because this could reduce functionally available oligonucleotides. However, perhaps transient self-hybridization would be enough to protect the 3′ end from degradation without losing efficacy? In theory, careful selection of nucleotide composition, chemical modifications, and length should be able to produce a segment with such transient self-hybridization.
With these ideas in mind, I developed a completely new antisense design: Cyclic structured oligonucleotides (CSO) containing two domains, a functional domain and a circularization domain to protect the 3′ end (Fig. 4). This circularization domain is linked to the functional domain through a 5′-5′ linkage and is complementary to the 3′ end of the functional domain, such that the CSO forms a transient cyclic structure with only a short overlap [122]. When the exposed single-stranded functional domain of the CSO hybridizes to a target RNA, the higher duplex affinity and proceeding zip-like hybridization will displace and thus release the circularization domain. Since this newly exposed 3′ end consists of PO-DNA without any modification, exonucleases will easily degrade it.
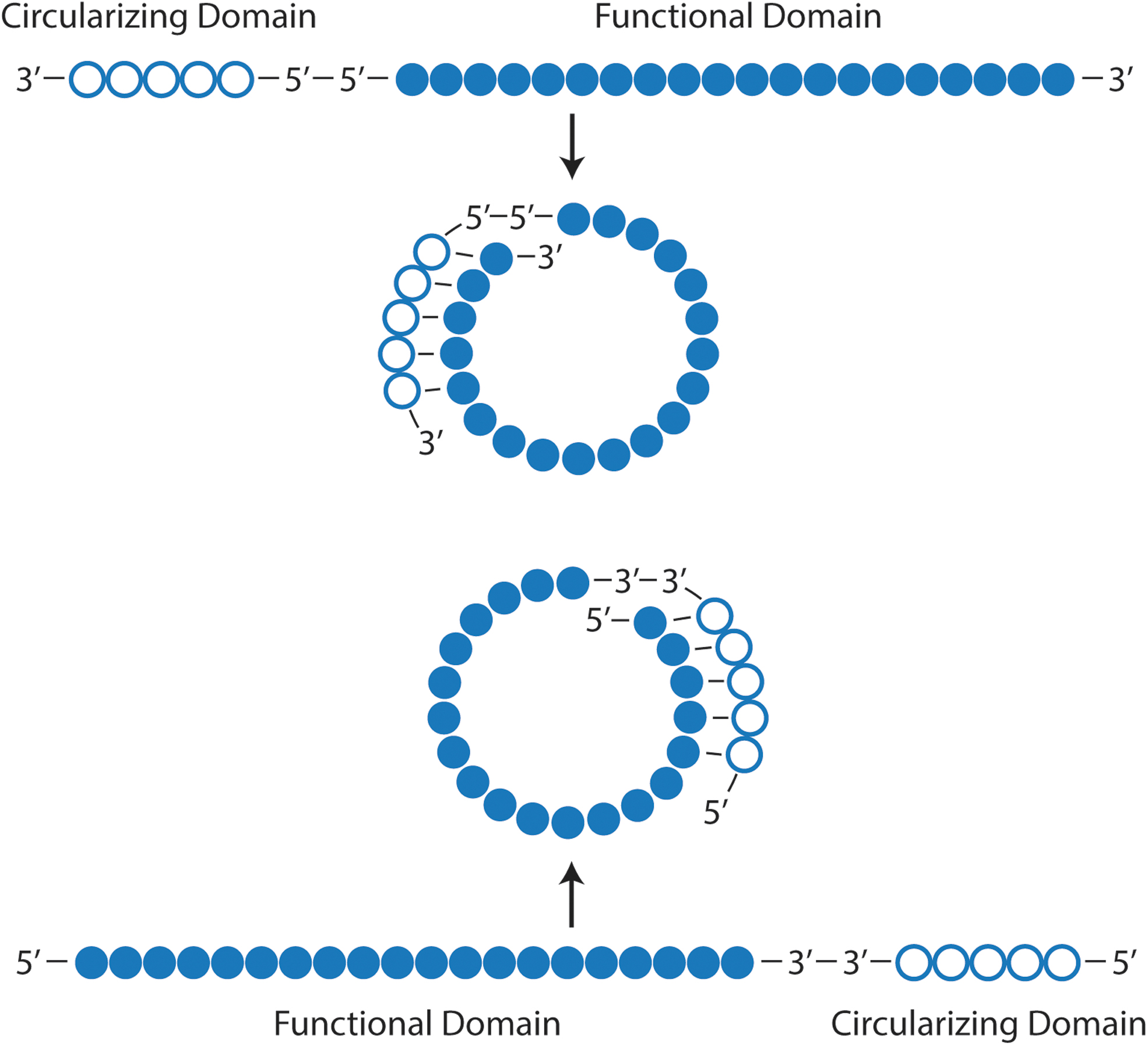
Design of CSO. There are two types of designs—first, where the functional domain is linked to a circularization domain by 3′-3′ links, and the second, where the two domains are linked by 5′-5′ linkage. Both designs form the same cyclic structure, except that the first contains two 5′ ends, and the second contains two 3′ ends. CSO, cyclic structured oligonucleotides.
The functional domain could be any antisense design composed of PS-DNA, PS-2′RNA, or a combination thereof, including gapmers or splitmers or even other modifications of varying length. Really, any nucleic acid molecule designed to hybridize to targeted RNA and recruit proteins involved in processes such as sequence editing, splice modulation, or upregulation of gene expression could be placed as the functional domain. Furthermore, any otherwise beneficial chemical modification we had previously discarded because of a reduction in protein binding and therefore delivery is an option again.
To test my theories, I designed a CSO with a splitmer as the functional domain and evaluated efficacy compared to a PS-DNA, a gapmer, and a splitmer in cells. All showed dose-dependent knockdown of the target gene, PNPLA3. Still, the splitmer demonstrated improved efficacy over the gapmer, which in turn was more potent than the PS-DNA (Fig. 5). However, the CSO design improved antisense efficacy by more than 20-fold over the splitmer and similar impressive improvements in potency have been seen with a number of antisense sequences studied so far (Fig. 5). I really had not expected that much difference!

Comparative efficacy of various antisense designs with sequences as shown. Antisense sequence is complementary to PNPLA3 mRNA. The sequence in blue is PO-DNA, in green is PS-DNA, and in orange is PS-2′-O-methoxyethoxy. In gapmer antisense, four nucleotides on both 3′ end and 5′ end are PS-2′-O-methoxyethoxy. Splitmer 1 and 2 contain 10 and 11-mer PS-DNA segments, respectively, and the rest of the sequence is PS-2′O-methoxyethoxy. Cyclic 1 and 2 designs have the same sequences as splitmer 1 and 2 in the functional domain, but with an additional 6-mer PO-DNA circularization domain. Dose–response for PNPLA3 downregulation is shown. Candidates were transfected into HepG2 cells, and after 24 h, target knockdown was assessed by qPCR compared to control POLR2A. A significant increase in potency and level of knockdown is observed with splitmer and splitmer cyclic structured antisense. qPCR, quantitative polymerase chain reaction.
To me, this remarkable increase in potency suggests that only a small fraction of gapmer is utilized for target RNA knockdown, and the rest is sequestered into, as yet undefined, cellular compartments and thus not available. Unspecific protein binding due to the anionic nature of PS linkages appears to be the likely culprit there. In my opinion, CSOs manifest reduced polyanionic characteristics and immune activation since a number of the PS linkages are masked by the duplex between the circularization and functional domains. The CSO design also allows for further reduction of PS linkages in the functional domain, which may decrease unspecific protein binding and thus increase potency even further.
In addition, availability of endogenous RNase H is clearly not a limiting factor for antisense functionality, otherwise such improvement in potency over gapmers would be impossible. To better understand the reasons for this tremendous increase, I am now investigating the contribution that various aspects make (use of the 3′ and 5′ ends, protein binding, interaction with PRRs, endosomal escape, RNase H-mediated cleavage, elimination from the cellular compartment, etc.).
A corresponding structure, but with a 3′-3′ link between the circularization and functional domains instead of a 5′-5′ link, did not show similar improvements in antisense efficacy. As the circularization domain is complementary to the 5′ end of the functional domain and thus the structure lacks accessible 3′ ends, this is congruent with our results from antisense oligonucleotides lacking 3′ ends. These findings demonstrate that we were focusing on the wrong ends of antisense oligonucleotides 25 years ago and opening up previously abandoned roads.
Returning to our work on immunostimulatory oligonucleotides, I wondered if a CSO design could also increase potency. So, I created 3′-3′- and 5′-5′-linked CSOs in which the functional domain contains immune stimulatory PS-DNA. As expected from our earlier research, the 3′-3′-linked CSO stimulated immune responses, while the 5′-5′-linked version mitigated them (Fig. 6) [122]. The increased immune stimulation with the 3′-3′ CSO was significantly higher than with the linear parental PS-DNA sequence. I am currently investigating a number of 3′-3′-linked CSOs of varying length and base composition, containing PS-DNA and PS-RNA as immune modifiers for PRRs and, ultimately, for immunotherapy.
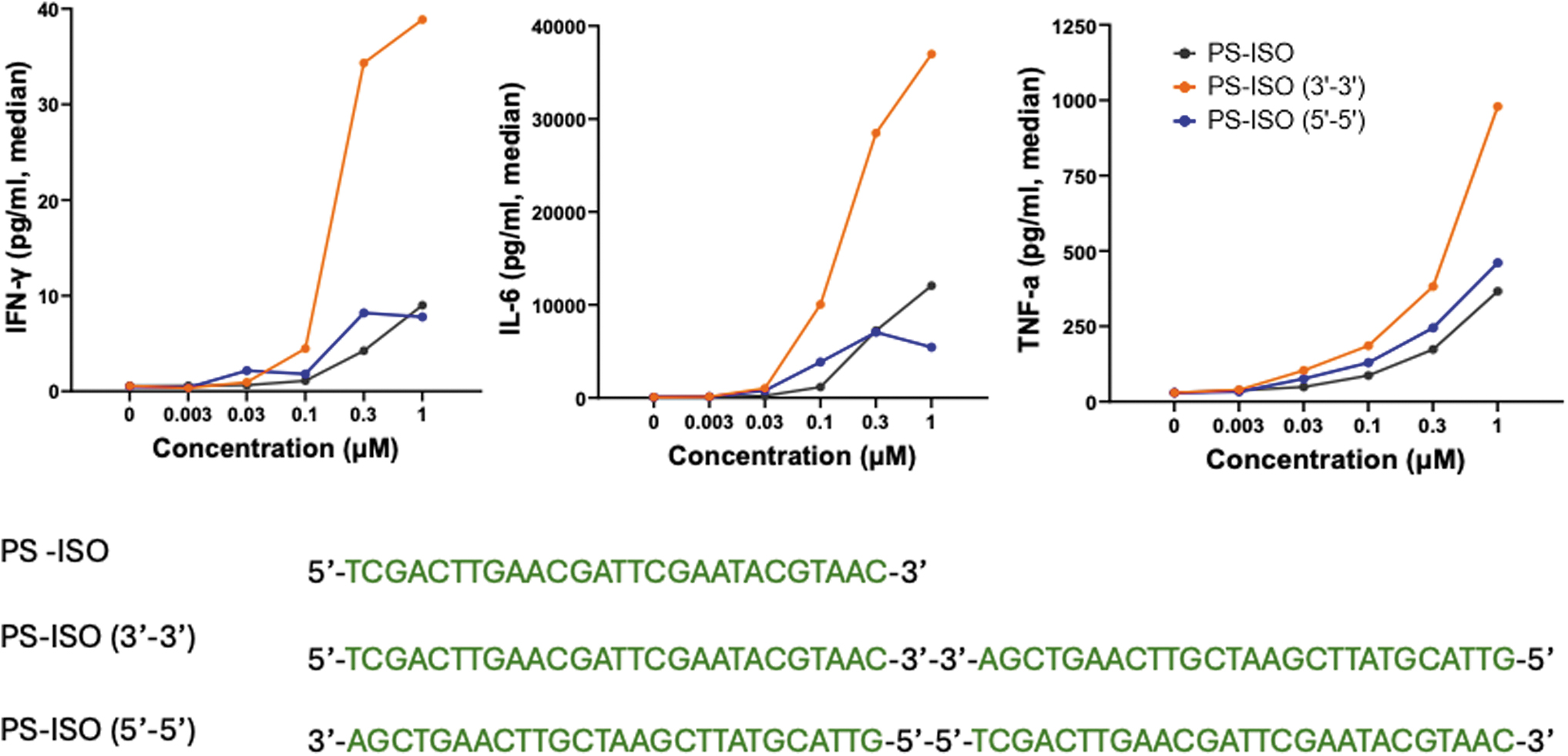
Contrasting characteristics of 3′-3′- and 5′-5′-linked CSO. The functional domain of both candidates consists of the same sequence, a CpG rich that acts as a TLR9 agonist. However, the immunostimulatory characteristics are very different. The immunostimulatory property is mitigated in CSO in which the 5′ end is inaccessible. Cyclic structure oligonucleotides with a 3′-3′ link are significantly more immunostimulatory than a linear PS-DNA. Data shown in this study are from evaluating these candidates in mouse splenocytes, treated for 24 h, and a Multiplexed MSD-based kit measured levels of cytokines. TLR, toll-like receptor.
Summary
Our quest to fulfill Paul's initial vision of making antisense oligonucleotides into viable therapeutics has become a reality. During that quest, we followed many promising roads, and, in the end, we arrived at our goal of successfully treating previously untreatable diseases. However, along the way, we turned away from many roads that seemed to hold no promise with what we knew then. Nowadays, the quest is no longer to make successful oligonucleotide therapeutics, but to make these treatments better and more widely applicable than they are. With more knowledge and deeper understanding, exploring those abandoned roads is a great time. Perhaps such exploration can advance not only the field of oligonucleotide therapeutics but also related fields where oligonucleotides are part of the therapeutic, such as RNA, gene, and prime editing.
All in all, my retirement in 2017 now seems rather fortuitous to me—it allowed me the time and mental space to THINK about my research, and I cannot wait to see where these new roads will take us.
Footnotes
Acknowledgments
I am indebted to my mentors, Mike Gait, Paul Zamecnik, Dan Brown, and Har Gobind Khorana, for their guidance, advice, discussions, and encouragement during this work. I am grateful to Petra Disterer for her expert editorial assistance and Beth Mellor for the figures. I express my gratitude to Greg Fredericks and Beau Carlson of Alloy Therapeutics for generating data in Figs. 4 and ![]() using established assays.
using established assays.
Author Disclosure Statement
S.A. is the founder of ARNAY Sciences LLC and inventor of the splitmer and cyclic antisense patent applications described in this article.
Funding Information
No external funding was received.