
Abstract
Despite high specificity and potency, small interfering RNA (siRNA)-based therapeutics have been limited by their poor biostability and intracellular penetration. Thus, effective nanocarriers that can protect and efficiently deliver siRNA to target cells in vivo are needed. Here we report on the efficiency of imidazole-modified chitosan (chitosan-imidazole-4-acetic acid [IAA])-siRNA nanoparticles to mediate gene silencing after administration via either intravenous (i.v.) or intranasal (i.n.) routes. Poly(ethylene glycol) (PEG)ylated nanoparticles for i.v. delivery demonstrated significant knockdown of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) enzyme in both lung and liver at as low as 1 mg/kg siRNA dose. In addition, the efficient, dose-dependent silencing of apolipoprotein B in the liver was also shown. For i.n. delivery, significant silencing of GAPDH protein expression was seen in the lungs with only 0.5 mg/kg/day siRNA delivered over 3 consecutive days. In summary, imidazole-modified chitosan-IAA nanoparticles are potentially effective carriers for siRNA delivery.
Introduction
However, like other nucleic acid-based therapies, siRNA-based approaches are hindered by serum instability of the RNA molecules, poor intracellular penetration of naked siRNA, and lack of inherent tropism. Therefore, only minimal success has been demonstrated with the use of naked siRNA in vivo, showing significant gene silencing only when directly administered to the tissue of interest at a very high dose leading to potential off-target silencing (Lewis et al., 2002; McCaffrey et al., 2002; Lewis and Wolff, 2005; Hickerson et al., 2008). Direct chemical modifications of siRNA have been reported for enhancing stability, cellular uptake, and targeting; however, the majority of these chemical modifications only provide minimal improvement on overall delivery efficiency (Amarzguioui et al., 2003; Elmen et al., 2005; Liao and Wang, 2005). To overcome these limitations, both viral and nonviral delivery vectors are being extensively explored for enhanced delivery of siRNA (Ramon et al., 2008).
Although viral vectors have proved to be highly efficient nucleic acid carriers, inherent safety concerns such as cytotoxicity, oncogenicity, and immunogenicity limit their potential use. In vivo lipid-based vehicles have demonstrated efficient delivery for siRNA; however, in addition to systemic cytotoxicity concerns, most have been target specific with predominant uptake in the liver, thus providing limited versatility (Morrissey et al., 2005; Zimmermann et al., 2006). Conjugation of peptides such as TAT(48-60) to siRNA has also shown the capability to improve gene silencing effects (Moschos et al., 2007). In recent years, polymer-based carriers for siRNA have garnered significant attention due to their potential biocompatibility, modifiability, and ease of production. Specific focus has been placed on cationic polymers, which form nanoscale polyplexes with siRNA, thus potentially minimizing serum degradation and allowing for endocytic intracellular uptake.
The polysaccharide chitosan and its derivatives have been an area of focus in nucleic acid delivery due to its inherent cationic nature, biocompatibility, and mucoadhesive properties (Roy et al., 1999; Mao et al., 2001). The predominant focus for use of chitosan and its derivatives as nonviral delivery agents has been for the delivery of pDNA and oligodeoxynucleotides (Roy et al., 1999, 2003; Gao et al., 2005), whereas recent research has focused on chitosan as a delivery vehicle for siRNA (Howard et al., 2006; Katas and Alpar, 2006; Pille et al., 2006; de Martimprey et al., 2008). However, although highly promising, chitosan is limited as a pharmaceutical delivery vehicle due to poor solubility above pH 6.5, low buffering at endosomal and physiological pH (5.5–7.4), and poor cytoplasmic dissociation kinetics, resulting in in vitro transfection efficiencies that are magnitudes lower than other polymeric and lipid-based delivery vectors.
Development of chitosan derivatives to enhance both in vitro and in vivo transfection has mostly focused on modification of the free primary amines. These include trimethylation for enhanced solubility (Florea et al., 2006), polyethylenimine conjugation for enhanced buffering (Jiang et al., 2007), poly(ethylene glycol) (PEG)ylation for serum stability (Mao et al., 2001), thiolation for enhanced mucoadhesive properties (Bernkop-Schnurch et al., 2004), and galactosylation for targeting to the liver (Park et al., 2001). Although these modifications demonstrated some improvements, none have addressed the aforementioned limitations in a comprehensive manner to produce a safe yet efficient systemic or mucosal siRNA carrier using chitosan.
Recently, we reported an imidazole-functionalized chitosan for pDNA and siRNA delivery, demonstrating significant increase of in vitro transgene expression as well as highly efficient in vitro gene silencing compared with nonmodified chitosan (Ghosn et al., 2008). Enhanced transfection efficacy was largely attributed to the introduction of secondary and tertiary amines to the polymer backbone as well as increased solubility at physiological pH (Pack et al., 2000) without altering the cyto-compatibility of the polymer. Here we report in vivo evaluation of this imidazole-modified chitosan (chitosan-imidazole-4-acetic acid [IAA]) and its potential as a delivery vehicle for siRNA to liver and lungs via multiple routes of administration. Our results indicate that chitosan-IAA is highly efficient in delivering bioactive siRNA in vivo via both the intravenous (i.v.) and intranasal (i.n.) routes. i.v. administration with PEGylated siRNA-chitosan-IAA nanoparticles resulted in up to 50% average gene silencing in the lungs and up to 40% silencing in the liver in a dose-dependent manner, with significant gene silencing even at a low 1 mg/kg dose. On the other hand, i.n. administrations of non-PEGylated nanoparticles lead to, on average, 50% silencing of the target gene in the lungs.
Materials and Methods
Materials
Chitosan (hydrochloride salt, Protasan UP CL113, molecular weight [Mw] = 130,000 Da, degree of deacetylation = 86%) was purchased from Novamatrix. IAA monohydrochloride, and MES Hydrate was purchased from Acros Organics. 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) hydrochloride and Snakeskin pleated dialysis tubing (10,000 molecular weight cut-off [MWCO]) were both purchased from Pierce Biotechnology. Modified with PEG-succinimidyl valerate (mPEG-SVA, Mw = 5000 Da) was purchased from Laysan Bio. Silencer® Select anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) siRNA, Silencer Select apolipoprotein B (ApoB) siRNA, Silencer Negative Control siRNA, and siPORT Amine were provided by Ambion Products, Life Technologies Corporation.
Mice
Female BALB/C and C57BL/6J mice for in vivo studies were purchased from Jackson Laboratory and housed at the Animal Resource Center, University of Texas, at Austin. Mice were 6–13 weeks old during treatment with nanoparticles. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas at Austin. Animals were provided care in accordance with procedures described in Animal Care and Use Handbook (University of Texas at Austin) and Principles of Laboratory Animal Care (NIH publication #85-23, revised in 1985).
Synthesis of imidazole-modified chitosan
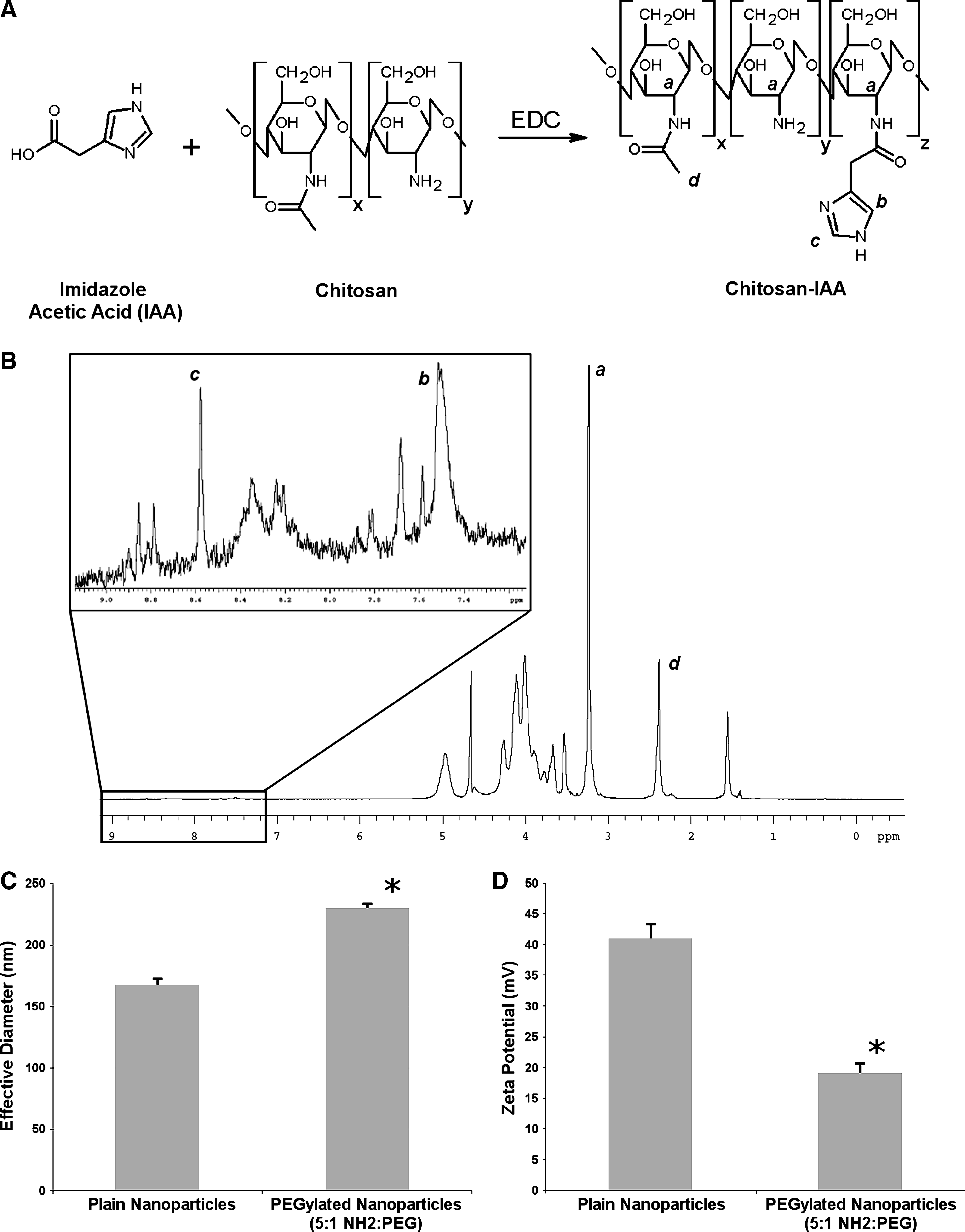
Conjugation of IAA monohydrochloride to the primary amines of chitosan was performed via carbodiimide chemistry, as previously described (Ghosn et al., 2008) with slight modification. Briefly, the ratio of IAA to the number of primary amines in chitosan was adjusted to provide variable degrees of modification. A schematic for this reaction and the final chemical structure of chitosan-IAA are shown in Fig. 1A. Chitosan (1% w/v, 49.3 mM NH2) and imidazole acetic acid (2.0% w/v, 123 mM) solutions were prepared in 0.2 M MES buffer (pH 5.5) and stored on ice. Chitosan (100 mg, 493 μmol NH2, 10 mL) was mixed with IAA (40 mg, 246.5 μmol, ∼2.0 mL) and kept on ice. The combined solution was then used to dissolve 20 M excess of EDC (945 mg, 4.93 mmol) to IAA and immediately vortexed for 60 seconds to catalyze conjugation of IAA along the chitosan backbone. The final solution was left to react overnight on an end-over-end rotator at room temperature. Reaction volumes were then dialyzed for 24 hours using Snakeskin pleated dialysis tubing against 5 mM HCl for 2 hours (3 × ) and then against deionized water for 6 hours (3 × ). Samples were then lyophilized overnight. Degree of substitution was characterized via 1H NMR with samples prepared in D2O and measured at 70°C using a Varian i.n. OV-500 (500 MHz) spectrometer with spectra shown in units of ppm.
(
Nanoparticle synthesis for intranasal administration
Nanoparticle formulations for i.n. administration were prepared with siRNAs (Silencer Select GAPDH and Silencer Negative Control), as previously described (Ghosn et al., 2008) with slight modification. Nanoparticle synthesis was performed as follows: Chitosan and chitosan-IAA solutions (0.045%–0.065% w/v) in RNase-free sodium acetate (200 mM, pH 4.5) were heated to 55°C for 20 minutes. Solutions of siRNA (20 μg/mL) were prepared in RNase-free water and preheated to 55°C for 1 minute before nanoparticle synthesis. Equivolumes of chitosan or chitosan-IAA solutions were added drop-wise to the siRNA solutions at a nitrogen:phosphate (N/P) ratio of 50 while vortexing. Samples were then vortexed for 30 seconds, resulting in final volumes of 200 μL and allowed to incubate at room temperature for 30 minutes. After incubation, samples were combined and concentrated with Amicon Ultra centrifugal filtration units (Millipore, MWCO = 30,000) to provide siRNA doses of either 0.5, 1, or 2.0 mg/kg (mg siRNA/kg body weight) in 20 μL volumes. Samples were centrifuged in filters at 1500 g for up to 40 minutes to adequately concentrate doses. Nanoparticles were then immediately characterized or used for in vivo studies. Nanoparticle size distribution was determined by Dynamic Light Scattering using a ZetaPlus system (Brookhaven Instruments Corporation). Zeta potential (ζ) for the nanoparticles was also determined using the ZetaPlus system. Briefly, nanoparticle solutions in 1 mM KCl (pH 5.5) were prepared at 0.25 mg/mL concentrations of polymer and read at room temperature with each sample read 10 times. The N/P ratio for each formulation was calculated as the ratio of protonable amines within the chitosan or chitosan-IAA at pH 4.5 to the phosphates of the siRNA backbone.
Nanoparticle synthesis and PEGylation for intravenous administration
For intravenously administered nanoparticles, synthesis was performed with 50 μg/mL siRNA solutions with chitosan and chitosan-IAA solutions adjusted accordingly. Particles were otherwise prepared identically to those described for i.n. administration at N/P ratios of 50 and 40, for GAPDH and ApoB studies, respectively, with a PEG surface modification. A slightly lower N/P ratio was used for ApoB studies to reduce complications due to viscosity and potential toxicity of the formulations at the higher doses. After 20 minutes of incubation at room temperature, nanoparticles were surface modified with mPEG-SVA (Mw = 5000) to prevent in vivo nanoparticle aggregation and enhance serum stability. Briefly, 10 mL of nanoparticles was mixed with mPEG-SVA (5% w/v in phosphate-buffered saline [PBS], pH 7.4) and vortexed for 60 seconds. Samples were then incubated for 2 hours at room temperature in a molar ratio of 1:5 (PEG:NH2 in chitosan or chitosan-IAA). After incubation, samples were then concentrated in Amicon Ultra centrifugation filtration units to provide 200 μL volumes for each dose for in vivo studies. Nanoparticles were characterized as described for non-PEGylated nanoparticles.
Intranasal administration of chitosan-IAA nanoparticles to C57BL/6J and BALB/C mice
In vivo gene silencing of GAPDH, after i.n. administration of chitosan and chitosan-IAA/siRNA nanoparticles, was studied on naïve C57BL/6J or BALB/C mice. Mice were separated into groups for administration of various nanoparticle formulations to each group (n = 5). PBS and Silencer Negative Control siRNA were used as negative controls for each set of experiments. Mice were placed under anesthesia with isofluorane and were administered 20 μL (10 μL per nostril per mouse) of chitosan-siRNA, chitosan-IAA-siRNA, or siPORT Amine-siRNA (positive control) nanoparticles. For initial i.n. studies, mice were administered nanoparticle formulations at an siRNA dose of 0.5 mg/kg for each mouse. For multiple administration studies, mice were administered the same 0.5 mg/kg siRNA dose daily for 3 consecutive days, resulting in a total 1.5 mg/kg of siRNA/mouse. To evaluate the effect of variable single siRNA doses, nanoparticle formulations were administered as a single dose of 0.5, 1.0, or 2.0 mg/kg siRNA per mouse. In all studies, mice were sacrificed and whole lungs were harvested for analysis at 3 days after final nanoparticle administration. After immediate snap freezing, tissues were manually homogenized and processed to determine GAPDH protein or mRNA silencing, as later described.
Intravenous administration of PEGylated nanoparticles to BALB/C and C57BL/6J mice
Both BALB/C and C57BL/6J mice (ages ranging from 6 to 9 weeks) were studied for nanoparticle-mediated in vivo gene silencing of GAPDH and ApoB after i.v. administration. In each experiment, mice (n = 5 mice per group) were placed in a warm restrainer and intravenously administered with 200 μL of nanoparticle formulations (chitosan or chitosan-IAA) via tail vein injection at Silencer Select siRNA with a single 1 mg/kg siRNA dose per mouse for GAPDH silencing studies. For ApoB studies, mice were administered equal daily injections of either 0.5 or 1.5 mg/kg siRNA per mouse over 2 consecutive days, resulting in final doses of either 1 or 3 mg/kg siRNA/mouse. Before administration, nanoparticle solutions were thoroughly mixed to minimize potential aggregates. PBS and Silencer Negative Control siRNA were used as negative controls for each experiment. In this study, Silencer Select siRNAs used contained a locked nucleic acid modification (Koshkin et al., 1998). In all experiments, mice were treated with nanoparticles at either an N/P ratio of 40 (ApoB) or 50 (GAPDH). Three days after the final injection, treated animals were sacrificed and organs (lungs and/or liver) were harvested and either snap-frozen and stored at −70°C or placed into RNAlater solution on ice and stored for further analysis. Tissue samples were then homogenized and analyzed for either GAPDH protein levels or ApoB mRNA levels, as described in the later sections.
Analysis of GAPDH protein silencing in tissue samples
After harvesting, organs evaluated for GAPDH protein content were snap-frozen in liquid nitrogen and stored at −70°C. Tissues were then homogenized as previously described (Hanson et al., 1998) and analyzed using the KDalert GAPDH assay kit (Ambion) according to manufacturer's protocol to determine protein levels of GAPDH. Briefly, homogenized tissue samples were resuspended in KDalert Lysis buffer (1 mL for lungs and 5 mL for liver) and incubated at 4°C for 20 minutes. Ten microliters of diluted lysate (1:1000–1:10,000 dilutions) from each sample was then transferred to a fresh black 96-well plate, and 90 μL of KDalert master mix was added to each well. Fluorescence levels for each well were read immediately after addition of the master mix (λExc. = 560 nm and λEmm = 590 nm). Identical measurements were repeated after 4 minutes. GAPDH activity was determined by the difference of the 2 readings. Values were normalized to the total protein content (Micro BCA assay; Pierce Biotechnology) of the lysate sample.
RNA isolation and quantitative polymerase chain reaction analysis of gene expression in tissue samples
To determine ApoB or GAPDH silencing at mRNA levels, total RNA was isolated from tissue samples using mirVana™ PARIS™ Kit (Ambion) per manufacturer's protocol. After total RNA isolation, samples were resuspended in nuclease free water and treated for genomic DNA contamination with a Turbo DNA-free™ kit (Ambion). To ensure quality and determine the quantity of mRNA isolated, samples were analyzed on an ND-1000 spectrophotometer (Nanodrop). Complementary DNA was synthesized using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The reverse transcription was performed with the cycle as follows: initially at 25°C for 10 minutes followed by 37°C for 120 minutes, and completed at 85°C for 5 minutes to inactivate the reaction as per the manufacturer's protocol. Samples were then held at 4°C. cDNA samples were analyzed by qPCR to determine gene expression level using the TaqMan Universal PCR Master Mix and an ABI Prism® 7900HT Fast Real-Time PCR System (Applied Biosystems). TaqMan Primers for GAPDH, ApoB, and 18S (housekeeping gene) were from Applied Biosystems. qPCRs were performed using manufacturer's protocol. Briefly, reaction volumes were incubated at 95°C for 10 minutes to activate Taq polymerase followed by 40 cycles of 95°C for 15 seconds for the denaturation of duplexes, and 60°C for 1 minute to allow for elongation. The relative level of gene expression for each sample is calculated by the ΔΔCT method (Livak and Schmittgen, 2001). Threshold values (CT) for both the target gene (ApoB or GAPDH) and housekeeping gene (18S or GAPDH) were determined with the ABI Prism 7700 Sequence Detection System software. To determine gene silencing, CT values for each sample were first normalized by subtracting the CT values of the housekeeping genes from each sample CT. Gene expression levels were then determined by first determining the ΔΔCT values obtained by subtracting the normalized CT values for control groups (PBS) from the normalized CT values of each test group sample. To ensure primer and total RNA quality, negative controls were analyzed by running samples without templates. To ensure total RNA purity, samples were concurrently run during cDNA synthesis without the use of reverse transcriptase to ensure no genomic DNA contamination (Singh et al., 2008).
Statistical analysis
To ensure statistical significance, all experiments were performed with at least 3 samples per group (n = 3). Standard deviations were calculated for each group, and analysis of statistical significance was determined using either a 2-tailed Student's t-test or one-way analysis of variance with Tukey's test to compare between treatment groups. Significance was set for values where P < 0.05. For in vivo studies, the numbers of mice in each group were determined by use of power analysis targeting a difference in means of 20% with a standard deviation of 10%, using an α = 0.05 and a power of 0.80. This resulted in a sample size of 5 mice per group. All error bars represent standard deviation.
Results
Synthesis and characterization of imidazole-modified chitosan
IAA-modified chitosan (chitosan-IAA) was prepared using a 1-step carbodiimide reaction, as previously described (Ghosn et al., 2008). The chemical structure for this modified chitosan is shown in Fig. 1A. Degree of substitution for the modified polysaccharides was determined via 1H NMR by taking the ratio of the 2 imidazole proton peaks (∼7.2–7.8 and ∼8.0–8.8 ppm), represented by “b” and “c” in Fig. 1B and the integration of the peaks correlating to the C2 protons of the IAA-N-acetyl glucosamine, N-acetyl-glucosamine, and glucosamine subunits at ∼3.2–3.4 ppm (represented by “a” in Fig. 1B). Peak “d” within Fig. 1B represents the methyl groups of the N-acetylated C2 protons of the chitosan-IAA. Chitosan-IAA batches used for these studies were characterized as having an average of ∼2%–3% degree of substitution. This is equivalent to approximately, on average, 15–23 molecules of IAA per chitosan polymer chain. IAA-modified chitosans demonstrated increased solubility and buffering, as previously described (Ghosn et al., 2008).
Nanoparticle synthesis and characterization
Nanoparticles were synthesized by complex coacervation of nonmodified chitosan or chitosan-IAA with siRNA, as previously described (Ghosn et al., 2008). N/P ratios of 40 and 50 were used for particle synthesis. Nonmodified nanoparticles have been previously shown to range in effective diameter from 150 to 300 nm dependent on the N/P ratio used, and they demonstrated a general trend of decreasing size with increasing N/P ratio (Ghosn et al., 2008). PEGylated particles at an N/P of 50 demonstrated a slightly higher effective diameter of 230 nm as compared with non-PEGylated nanoparticles, which had an effective diameter of ∼168 nm, as shown in Fig. 1C. It was also noted that the mean effective diameter for the nonmodified nanoparticles was not drastically changed after concentration of the formulations. As shown in Fig. 1D, reduction in zeta potential was also noted for particles after PEGylation. Surface charges for nonmodified nanoparticles were +41.1 mV, whereas PEGylated nanoparticles demonstrated a surface charge of +19.1 mV.
Silencing of GAPDH in murine lungs and liver after intravenous delivery of PEGylated chitosan-IAA/siRNA nanoparticles
Although highly desirable, i.v. delivery of siRNA molecules has been limited by serum instability, lack of intracellular and tissue penetration, and lack of targeting. In vivo experiments were conducted to determine the capability of PEGylated, nontargeted chitosan-IAA/siRNA nanoparticles to induce gene silencing in the lungs and liver after the i.v. injection.
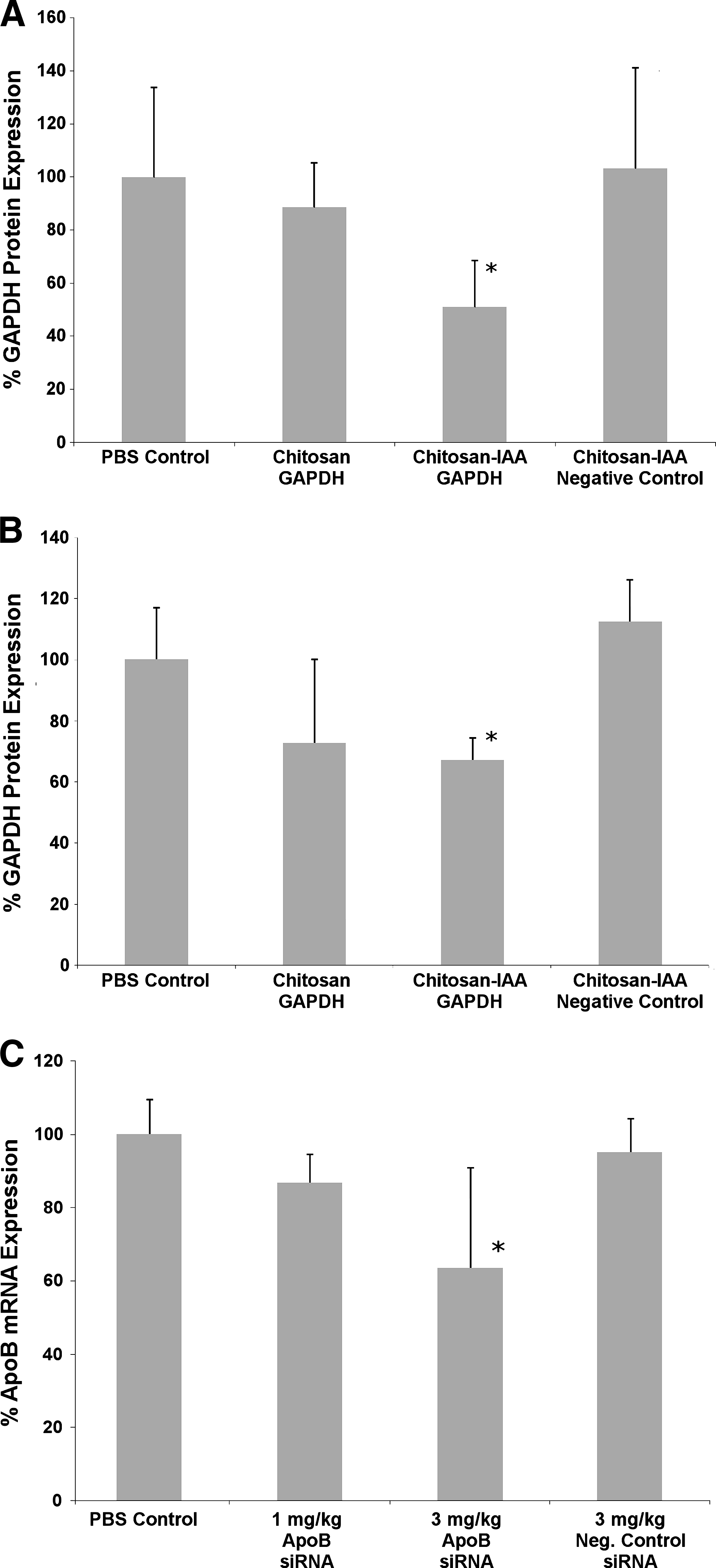
Female BALB/C mice (6–8 weeks of age) were injected intravenously via the tail vein with chitosan/siRNA and chitosan-IAA/siRNA (GAPDH) nanoparticles, and the lungs and liver were analyzed for GAPDH silencing. The earlier characterized (Ambion cross-species positive control that targets the human, mouse, and rat gene) hyper-potent GAPDH siRNA that was used in this study has been previously identified via extensive in vitro screening. To counteract potential serum-induced aggregation due to protein adsorption, PEG (mPEG-SVA) was conjugated to the nanoparticles at a ratio of 1:5 (PEG:NH2). Initial studies were performed by injection of nanoparticles at a concentration of 1 mg/kg siRNA to each mouse and N/P ratio of 50. Three days after the injection, mice were sacrificed, and whole lungs and liver were harvested. Analysis of GAPDH protein expression in the lungs demonstrated a significant gene silencing effect of up to ∼49% when siRNA was delivered using chitosan-IAA (Fig. 2A). Chitosan/siRNA nanoparticles mediated minimal gene silencing effect for GAPDH (∼11%) when administered via i.v. injection, producing an insignificant silencing effect as compared with control groups. Similar results were noted in C57BL/6J mice (6–9 weeks of age) after treatment with identical formulations.
Silencing of GAPDH protein (
In vivo gene silencing in the liver after i.v. administration of PEGylated nanoparticles was also evaluated. As shown in Fig. 2B, 3 days after the injection, chitosan-IAA nanoparticles demonstrated significant gene silencing in the whole liver at ∼33% when compared with the PBS control group. Although chitosan nanoparticles provided ∼27% average gene silencing effect after the injection, high variability resulted in no statistically significant difference when compared with the PBS control group. Similarly to previous studies, negative control siRNA delivered with chitosan-IAA nanoparticles demonstrated minimal gene silencing effect.
Dose-dependent gene silencing of ApoB in the liver after treatment with chitosan-IAA/siRNA nanoparticles
Chitosan-IAA/siRNA nanoparticles were studied to further examine the effect of variable dose on gene silencing of ApoB, a carrier molecule for low-density lipoproteins (LDL). Female BALB/C mice were intravenously injected with nanoparticles prepared from chitosan-IAA and either ApoB-targeted Silencer Select siRNA or a Negative Control siRNA (Ambion). Three ApoB siRNAs predicted by the Silencer Select algorithm were evaluated in vitro, and the sequence that induced maximal knockdown of the mRNA target at lowest concentration was selected for in vivo studies (data not shown). Particles were prepared at an N/P ratio of 40 to minimize viscosity issues at higher doses and surface mPEG-SVA to improve serum stability. Mice were injected with final doses of 1 and 3 mg/kg siRNA via 2 injections over 2 consecutive days (200 μL/dose) through the tail vein. As shown in Fig. 2C, 3 days after the final injection, gene expression levels determined by qPCR demonstrated gene silencing of ∼15% and ∼37% for 1 and 3 mg/kg siRNA doses, respectively. Mice that were administered a total 3 mg/kg siRNA dose demonstrated significant gene silencing of ApoB as compared with all control groups. Groups administered with 3 mg/kg doses of negative control siRNA nanoparticles demonstrated minimal to no silencing as expected.
Chitosan-IAA/siRNA-mediated silencing of GAPDH in lungs after intranasal administration
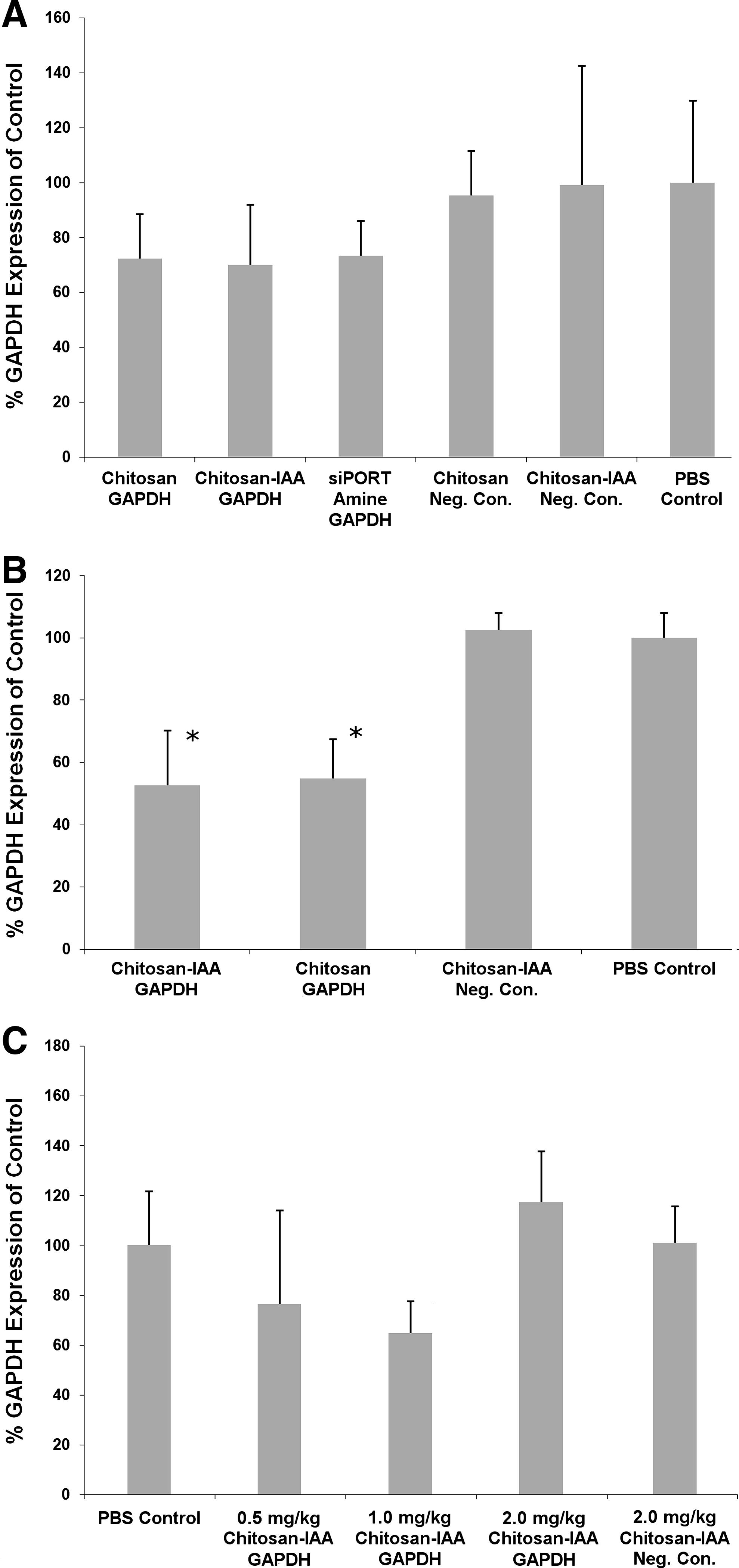
In vivo experiments were performed to compare the delivery efficiency of siRNA after i.n. administration with chitosan or chitosan-IAA/siRNA nanoparticles. Silencing of GAPDH expression in the lungs was evaluated at low siRNA doses (0.5 mg/kg single dose or 3 consecutive daily doses of 0.5 mg/kg). No observable adverse effects were noted in mice after administration of various nanoparticles formulations. Three days postadministration, treated mice were sacrificed and whole lungs were analyzed for GAPDH expression. After a single low-dose injection in C57BL/6J mice, some GAPDH knockdown was observed (Fig. 3A), although the differences between the experimental and control groups were not statistically significant. Mice treated with chitosan-IAA/siRNA, chitosan/siRNA, and siPORT Amine/siRNA all showed an average knockdown of 26%–30% in the whole lungs.
Silencing of GAPDH protein in lungs after intranasal administration of chitosan and chitosan-IAA/siRNA nanoparticles to C57BL/6 mice (
Since single low-dose administrations did not show statistically significant knockdown, we studied the use of multiple low-dose administrations to determine enhancement in gene silencing. Mice (C57BL/6J) were treated with an identical dose (0.5 mg/kg siRNA) daily for 3 consecutive days. Treated animals were sacrificed and lungs were collected 3 days after the final administration and analyzed for GAPDH protein content. Use of multiple doses demonstrated significant gene silencing (47%) for chitosan-IAA/siRNA nanoparticles as is shown in Fig. 3B. Similarly, chitosan/siRNA nanoparticles elucidated 45% silencing of GAPDH. There were no significant differences between the modified and nonmodified polysaccharide. The siPORT Amine nanoparticles demonstrated cytotoxicity in our initial multiple dose studies and, therefore, were not further used. In comparison, no silencing effect was noted for mice treated with negative control siRNA nanoparticles delivered with chitosan-IAA.
To study the effects of varying siRNA dosage on mRNA silencing after i.n. administration of chitosan-IAA/siRNA nanoparticles, siRNA doses of 0.5, 1.0, and 2.0 mg/kg of siRNA were evaluated in BALB/C mice. Higher doses of siRNA demonstrated no adverse affects on the mice after administration. As shown in Fig. 3C, GAPDH gene silencing (mRNA level) of up to 35% was demonstrated with a single dose when 1 mg/kg of GAPDH siRNA was delivered with chitosan-IAA. Low-dose samples (0.5 mg/kg) demonstrated similar silencing effects to earlier studies with gene silencing of ∼24%. The treatment of mice at the highest dose (2.0 mg/kg) for chitosan-IAA nanoparticles produced no silencing effect in mice when administered intranasally. As discussed below, this demonstrates a potential limitation, possibly due to the higher viscosity of the formulation, in the effective dosing range for chitosan-IAA-mediated siRNA for i.n. delivery.
Discussion
We have previously demonstrated effective enhancement of in vitro transfection efficiency with a chitosan-based delivery vector in which several primary amines of the polysaccharide were functionalized with an imidazole-containing molecule (imidazole acetic acid), thus introducing secondary and tertiary amine groups to the polymer backbone (Ghosn et al., 2008). We also showed enhanced buffering, increased solubility, and minimal cytotoxicity of the chitosan-IAA derivative at both endosomal and physiological pH levels. In the current study, we demonstrate that chitosan-IAA/siRNA nanoparticles provide efficient gene silencing in lungs and liver after administration via i.n. and i.v. routes.
A major challenge in nanoparticle-mediated siRNA delivery is the inherent “dilute” nature of the formulations that limits in vivo injection doses, especially for systemic delivery. It is critical that all such formulations be concentrated without particle aggregation. Chitosan-IAA/siRNA nanoparticles were successfully concentrated at the reported doses without significant aggregation. To increase serum stability for i.v. administration, PEG groups were successfully conjugated to the particle surface. PEGylation increased the effective hydrodynamic diameter of the nanoparticles (from an average of ∼170 to 230 nm) while decreasing zeta potential (from +41.1 to +19.1 mV). This is likely due to the reduction in free primary amines on the particle surface resulting from PEG attachment.
To demonstrate gene silencing efficacy after systemic administration, naïve BALB/C or C57BL/6J mice were injected via the tail vein with anti-GAPDH or anti-ApoB siRNA. We focused on analyzing the lungs and liver, because these organs are likely to take up majority of the nanoparticles and also have significant disease targets suitable for therapeutic use of siRNA. Further evaluation of biodistribution and knockdown in other organs is essential for future development and optimization of chitosan-IAA as an in vivo siRNA carrier. After administration of PEGylated chitosan-IAA nanoparticles, significant gene silencing was achieved in the whole lungs and liver (∼50% and ∼30% reduction in average GAPDH protein levels) even at a low dose (1 mg/kg siRNA). In comparison, minimal or highly variable knockdown was observed in mice treated with nonmodified chitosan nanoparticles.
ApoB is the predominant carrier protein for LDL within the blood, which is responsible for the transport of cholesterol throughout the tissues. Recent studies have demonstrated the therapeutic potential of siRNA in silencing ApoB for the treatment and prevention of coronary artery disease (Soutschek et al., 2004). i.v. delivery of cholesterol-conjugated anti-ApoB siRNA and the resultant silencing of the gene have been shown to lower total cholesterol (Soutschek et al., 2004). The potential of such treatments, however, is limited due to the high doses (≥20 mg/kg) necessary to achieve desired effects. In this study, we investigated the efficacy of chitosan-IAA in delivering anti-ApoB siRNA to the liver. Mice were treated intravenously with PEGylated chitosan-IAA/siRNA nanoparticles. Although minimal (∼13%) silencing of the ApoB gene expression was noted in the liver at 1 mg/kg siRNA dose, significant knockdown (∼37%) was seen at the 3 mg/kg dose. Some variability between samples was noted with several mice demonstrating up to ∼50%–70% knockdown of ApoB. This could be due to the inherent variability of ApoB levels in mice and its dependence on dietary intake of each mouse, which can be tested in future studies by testing serum ApoB levels in each treated animal before siRNA administration.
Although we have shown chitosan-IAA as an effective systemic carrier for siRNA, there may be limitations for its use, particularly at higher doses. At high doses, increase in viscosity after particle concentration may cause aggregation, especially at low PEG modification efficiencies. Thus, PEGylation plays a critical role in reducing potential toxicity due to particle aggregation after systemic administration. Based on these issues, it may be optimal to administer repeated low doses of the nanoparticles (1–2 mg/kg siRNA) over several days through the i.v. route rather than through large single doses. Further optimization of N/P ratio in future studies may also provide means to reduce potential aggregation issues.
Administration of therapeutic agents intranasally provides a potential delivery route to the lungs and upper airways, while effectively circumventing hepatic first-pass clearance. The i.n. route provides opportunity for exploitation of the inherent mucoadhesive properties noted with chitosan and its derivatives and provides a localized treatment, thus minimizing potential systemic side effects. To investigate the use of chitosan-IAA for i.n. delivery of siRNA, naïve C57BL/6J or BALB/C mice were intranasally administered nanoparticle formulations at low siRNA doses. Our results demonstrated effective gene silencing (up to 30%) in the entire lungs. Further, treatment of mice with multiple low doses (0.5 mg/kg) over 3 consecutive days provided significant silencing (∼52%) at a lower total dose than previously demonstrated with nonmodified chitosan/siRNA nanoparticles (Howard et al., 2006).
The study of polymeric and lipid-based delivery vehicles for i.n. or intratracheal siRNA delivery has increased over the past 5 years. The use of the lipid-based and natural surfactant Infasurf has been shown to effectively silence GAPDH protein expression within the lungs at similar doses to our study (Massaro et al., 2004). The use of TransIT TKO, a cationic polymer/lipid combination, has also demonstrated significant enhancement of siRNA delivery and silencing effect after i.n. administration reducing target viral titer by up to 99%, however, at siRNA doses 7-fold greater than our studies (Bitko et al., 2005).
Although chitosan-IAA/siRNA nanoparticles demonstrated significant in vivo gene silencing in the whole lung after i.n. administration, they showed no significant enhancement in silencing efficacy when compared with nonmodified chitosan/siRNA particles. This may be attributed to a reduction in mucoadhesiveness, which has recently been directly linked to the degree of deacetylation of chitosan (Sogias et al., 2008). Further, the higher viscosity of chitosan-IAA formulations may lead to a reduced amount of nanoparticles reaching the lungs after i.n. administration. An increase in gene silencing effect was noted from 0.5 to 1 mg/kg siRNA doses, whereas a loss of gene silencing effect was noted at the highest nanoparticle dosage and concentration, thus providing the possibility of minimal penetration into the lungs with higher viscosity formulations at higher concentrations. Further optimization of degree of substitution of imidazole acetic acid to chitosan for i.n. delivery is needed. Nevertheless, IAA-modified chitosan, which exhibits enhanced solubility and pH robustness compared with the nonmodified polymer, provides distinct advantages in pharmaceutical processing.
Although our results indicate the potential of chitosan-IAA as an in vivo carrier for siRNA, further evaluation, in terms of efficacy and safety of the nanoparticles, especially at high doses of siRNA and chitosan-IAA, is necessary to determine optimal properties and potential pitfalls. Biodistribution of the nanoparticles after both i.v. and i.n. administration would provide knowledge of the potential tissue targets for this system. Variation in the size of PEG molecules used for surface modification may provide improved uptake into various organs and circulation time and reduce potential aggregation-induced toxicity. Introduction of targeting moieties may also provide enhanced organ-specific uptake of the nanoparticles. Future work will aim at optimizing the chitosan-IAA/siRNA delivery system and determine its efficacy and detailed safety profile in therapeutic models via several routes of administration.
Conclusion
In conclusion, we have demonstrated the effective use of chitosan-IAA as an in vivo delivery vehicle for siRNA via both local (i.n.) and systemic (i.v.) delivery routes and shown significant enhancement of siRNA-mediated gene silencing in both the liver and lungs. Further optimization of formulation parameters, biodistribution, and safety profile are needed for organ-specific in vivo delivery of siRNA.
Footnotes
Acknowledgments
We would like to thank the University of Texas at Austin Animal Resource Center and its staff for their gracious support and helpful insight. We would also like to thank Dr. Nathalie Guimard for help with NMR data analysis and critical discussions. This work was partially supported by an NSF IGERT fellowship (B.G.).
Author Disclosure Statement
No competing financial interests exist.