
Abstract
RNA interference has proven to be a powerful tool to inhibit viruses. For the prevention of viral escape, multiple short hairpin RNAs (shRNAs) will have to be employed. This article describes a rapid procedure for the generation of shRNA expression cassettes by parallel cloning as well as a simple strategy for the combination of selected units. After delivery of the shRNA expression cassettes with adeno-associated virus vectors, inhibition of echovirus 30 as well as silencing of an important cellular cofactor of virus replication were achieved. The procedure has the potential to be generally applicable for silencing of multiple endogenous targets or viruses.
Introduction
Due to the limitations of siRNA delivery and the need for long-term silencing to prevent viral spread, virus vector-mediated nuclear expression of shRNA is a promising strategy to treat or prevent viral infections. In recent years, several viral vector systems for shRNA delivery to different cell types have been developed and successfully applied for in vitro as well as for in vivo applications (summarized in Grimm and Kay, 2007). Vectors based on adeno-associated viruses (AAVs) are promising vehicles for the delivery of shRNA expression cassettes and allow potent transduction of different target cells and tissues. Conventional AAV vectors contain a monomeric vector genome that expresses the transgene at a comparatively low level. Expression reaches the maximal level ∼72 hours after transduction in cell culture and in vivo even after several weeks: these weaknesses have been overcome by the development of AAV vectors with self-complementary (sc) vector genomes enabling rapid onset of transgene expression at higher levels (McCarty et al., 2001). We, therefore, prefer this type of AAV vectors for the treatment of acute virus infections (Fechner et al., 2008). Importantly, no pathogenicity was found for wild-type AAVs and the frequency of genomic integration for this type of virus vector is extremely low.
Echovirus 30 (EV-30), which is one of the most frequently isolated viruses from aseptic meningitis outbreaks worldwide (Oberste et al., 1999; CDC, 2006), is a member of the genus Enterovirus in the family Picornaviridae. Enteroviruses are small nonenveloped viruses containing an RNA genome in positive orientation (Racaniello, 2007). The genome encodes a single polyprotein that is subsequently cleaved into structural and nonstructural proteins. One of the nonstructural proteins, which is required for replication, is the RNA-dependent RNA polymerase (RdRP). Due to the high degree of conservation of the RdRP protein throughout the Picornavirus family (Whitton et al., 2005), its RNA is an attractive target to prevent virus replication by RNAi. We have previously described an approach to design siRNAs with high potency against a virus-specific target gene. In addition, we showed that silencing of the decay-accelerating factor (DAF) prevents viral spread of EV-30 (Rothe et al., 2009).
In the present study, we provide a new strategy for the easy and straightforward generation of vectors expressing one or multiple shRNAs based on prevalidated siRNA sequences. Starting point is a primer extension reaction, which was described previously for the generation of a single shRNA-encoding sequence (McIntyre and Fanning, 2006). We now employed this method successfully to generate numerous shRNA expression cassettes by parallel cloning in a single experiment. The combination of shRNAs expressed from a single vector has been proposed as a measure to prevent viral escape (Schubert et al., 2005; ter Brake et al., 2008). Here, we describe a green fluorescent protein (GFP) reporter assay for rapid testing of shRNAs and the simple combination of highly efficient shRNA expression cassettes by choosing compatible restriction sites (Xu et al., 2009) leading to vectors with multiple expression cassettes. Finally, AAV vectors for the delivery of these constructs were generated, and the silencing activity of each shRNA as well as the antiviral potency of the double-expression vector were demonstrated.
Materials and Methods
Cell culture and transfection
Rhabdomyosarcoma (RD), HeLa, and HEK293FT cells (Invitrogen) were grown as previously described (Rothe et al., 2009) except that the glucose content of Dulbecco's modified Eagle's medium for HEK293FT cells was increased to 4.5 g/L. For GFP reporter assays, RD cells were seeded at a density of 1.5 × 105 cells per well; for GFP reporter assays of shRNA double-expression plasmids, HeLa cells were seeded at a density of 1 × 105 cells per well, both in 24-well plates in a volume of 500 μL medium without antibiotics. One day after seeding, cells were cotransfected with 400 ng of the GFP reporter plasmid and the indicated amount of the shRNA expression vector using 1.5 μL Lipofectamine™ 2000 (Invitrogen) per well, according to the manufacturer's instructions.
Oligonucleotides and vectors
DNA oligonucleotides for polymerase chain reaction and for cloning procedures were purchased from Tib Molbiol and are summarized in Table 1. The pSilencer 2.1-U6 neo vector and the corresponding negative control vector that expresses a scrambled control shRNA were purchased from Ambion (now part of Applied Biosystems).
DNA sequences encoding the sense and antisense strand of the shRNAs are marked in bold. It should be noted that the loop of the shRNA for which the standard sequence recommended by the manufacturer was used can be expected to form 2 additional base pairs.
Abbreviations: shRdRP, short hairpin RNA-dependent RNA polymerase; shRNAs, short hairpin RNAs.
Primer extension
The universal primer (20 pmol) was annealed to the pool of single-stranded DNA (ssDNA) oligo templates (50 pmol each) by denaturing at 95°C for 2 minutes and cooling down slowly to room temperature. Next, 5 U of polymerase Phi29 (New England Biolabs) was added to the annealing reaction, which was supplemented with 1×Phi29 polymerase buffer, 200 μg/mL bovine serum albumin, and 0.2 mM dNTPs (each) to a final volume of 20 μL. The extension reaction was carried out at 30°C for 10 minutes and Phi29 polymerase was then inactivated by denaturing at 65°C for 10 minutes. The resulting double-stranded DNA (dsDNA) pool was subsequently incubated with BamHI and HindIII at 37°C for 20 hours and then cloned into pSilencer 2.1-U6 neo vector, which had been opened with the same restriction enzymes.
Cloning procedures
Cloning of the cDNA of RdRP of EV-30 into a GFP reporter plasmid was described previously (Rothe et al., 2009). The original shuttle plasmid for AAV vector production (pscAAV-GFP; Fechner et al., 2008) was modified by replacing the GFP expression cassette (containing cytomegalovirus [CMV] promoter, GFP, and bovine growth hormone polyadenylation signal sequence) by a stuffer sequence of 657 bp via NotI and HindIII sites. The resulting plasmid still contains the shRNA expression cassette of pscAAV-GFP (murine U6 promotor and CoxB3 RdRP2-shRNA), which was subsequently replaced by shRNA expression cassettes targeting RdRP of EV-30 and/or DAF via EcoRI and HindIII sites.
AAV vector production and quantification
For the production of AAV vectors, a genome with a mutation that leads to the formation of double-stranded vectors was used. In addition to the shRNA expression cassettes, an irrelevant stuffer sequence of 657 bp that will not be transcribed was included between the inverted terminal repeats, to keep the vector genome within a certain size range. AAV vectors were produced using a common protocol (GRIMM, 2002), but cotransfection of HEK293FT cells with the AAV shuttle plasmids containing the shRNA expression cassettes and a packaging plasmid (pDG) was performed at a ratio of 1:3 using 2.5 μL Lipofectamine 2000 per μg DNA. The produced AAV vectors were purified and concentrated on an iodixanol density gradient according to a previously published protocol (Zolotukhin et al., 1999), except for the use of 54% instead of 60% iodixanol as the highest concentration and the centrifugation step using a Type 70 Ti rotor (Beckmann) at 246,700 g for 2 hours at 18°C. Virus vector concentration was subsequently quantified using a quantitative polymerase chain reaction-based method (Veldwijk et al., 2002). Comparable titers were obtained for all vectors; that is, sequence and number of shRNA expression cassettes did not influence encapsidation or purification.
Transduction
HeLa cells were seeded in 24-well plates at a density of 1 × 105 cells per well in a volume of 500 μL medium. The next day, cells were transduced with either 1000 or 5000 vector genomes per seeded cell (vg/c).
Western blot
Western blots for GFP and actin were performed as described previously (Rothe et al., 2009). For the detection of endogenously expressed DAF, cells were harvested 3 days after transduction and proteins were separated on a 10% (w/v) sodium dodecyl sulfate-polyacrylamide gel. Membranes were then incubated with rabbit DAF (CD55) antiserum H-319 (1:1000; Santa Cruz Biotechnology Inc.).
Cell viability assay
RD cells were seeded in 96-well plates at a density of 1 × 104 cells per well in a volume of 100 μL medium. The next day, cells were transduced with either 1000 or 5000 vg/c. Three days after transduction, cells were infected with EV-30 (Bastianni, ATCC no. VR-322) at a multiplicity of infection of 0.1 plaque forming units/cell. Cell viability was determined 48 hours postinfection using the Cell Proliferation Kit II (Roche), following the manufacturer's instructions.
Results
Parallel cloning of shRNA expression cassettes
In a recent study, we provided a systematic approach to design siRNAs with high potency against a virus-specific target gene (Rothe et al., 2009). As a result, 5 siRNAs were obtained that are potent inhibitors of the RdRP of EV-30 in both reporter and virus assays. For long-term silencing of viral gene expression and for potential applications in gene therapeutic approaches, the use of viral shRNA expression vectors would be advantageous. The translation of siRNAs into shRNA coding sequences, however, still remains a challenging task. To date, little is known about factors that influence the efficacy of shRNAs designed on the basis of corresponding siRNA sequences. Therefore, we established a rapid and inexpensive parallel cloning procedure to simultaneously generate several shRNA expressing vectors.
As a first step, ssDNA template oligonucleotides were designed to encode the sense and antisense strand of selected siRNAs against the RdRP and a constant loop (Fig. 1a). For subsequent cloning into pSilencer 2.1-U6 neo, all constructs were flanked by a BamHI restriction site on the 5′ end and a HindIII restriction site on the 3′ end. For the option to generate double (or multi)-expression cassettes, an MfeI restriction site was inserted upstream of the HindIII site. A universal extension primer was directed against the invariant 3′ end of all ssDNA templates (Fig. 1a). For the generation of the dsDNA, 6 different ssDNA templates were pooled and incubated with universal primer and the polymerase Phi29. This DNA polymerase is known to combine high efficiency in extension reactions with reduced error rates as compared to the Taq polymerase (McIntyre and Fanning, 2006). The resulting dsDNA pool was processed with HindIII and BamHI, ligated into pSilencer 2.1-U6 neo and subsequently transformed in Escherichia coli. Randomly selected clones were sequenced. Approximately two-thirds of the clones exhibited the correct sequence (Table 2), while the other clones contained deletions or mutations. For 5 out of the 6 shRNA-encoding plasmids, correct clones were obtained. A closer inspection revealed that the DNA sequence encoding shRdRP6 harbors a HindIII restriction site so that only truncated sequences could be obtained.

Method for simultaneous cloning of several shRNA-encoding sequences into pSilencer plasmids. (
Subsequently, the efficacy of the shRNA expression plasmids shRdRP1 to shRdRP5 was evaluated. To this end, the cDNA of the RdRP of EV-30 was cloned downstream of GFP (Fig. 1b). Reporter constructs were cotransfected with each of the 5 shRNA expression vectors. As shown in the Western blot (Fig. 1c) shRdRP5 greatly reduced expression of GFP, while shRdRP1-4 resulted in partial silencing only.
Building block principle to generate multiple expression vectors
Viruses can escape long-term RNAi-mediated silencing by the selection of escape mutants (Boden et al., 2003; Das et al., 2004; Wilson and Richardson, 2005). As a countermeasure, the combination of siRNAs targeting (conserved) regions of the virus genome or host cell factors that are essential for virus entry can prevent the emergence of resistant viruses. For the generation of double (or multiple) shRNA-expressing vectors, we used the additional MfeI restriction site that was inserted downstream of the shRNA-encoding sequence (Fig. 1a). Restriction with MfeI yields an overhang that is compatible with the overhang generated by EcoRI (Fig. 2a). The sequence obtained by ligation is no longer recognized by MfeI or EcoRI.

Building block principle. The insertion of an MfeI restriction site allowed easy combination of shRNA expression cassettes. (
The first step of the building block principle (Fig. 2b) is a double digestion of the donor plasmid with the MfeI and EcoRI. The acceptor plasmid, which already contains at least 1 shRNA expression cassette, is linearized by EcoRI. Ligation of the shRNA expression cassette into the linearized acceptor plasmid yields the siRNA double-expression (SiDEx) vector. As a further development compared to our previous report on the generation of a SiDEx vector (Schubert et al., 2005), the new approach allows full flexibility with respect to the combination of selected shRNA expression cassettes. In addition, there is no limit to the number of shRNA expression cassettes that can be placed in a row. For example, addition of another shRNA expression cassette results in an siRNA triple expression vector.
For the present aim of inhibiting EV-30, we chose the combination of an shRNA against the viral genome and an shRNA targeting a host cell factor to minimize the risk of viral escape. We previously demonstrated that silencing of DAF by RNAi results in potent inhibition of EV-30 spread (Rothe et al., 2009). Thus, the expression cassettes shRdRP5 and shDAF2 were combined in a pSilencer plasmid (Fig. 2c). Only clones in which both shRNA expression cassettes were oriented in parallel direction were sequenced and used for further experiments. The orientation of the 2 shRNA expression cassettes could easily be tested by an EcoRI/HindIII restriction.
Reporter assays in analogy to the experiments described above revealed a concentration dependent and efficient inhibition of GFP (Fig. 2d), confirming that the silencing capacity was not reduced by the combination of 2 expression cassettes, each of which contained a U6 promoter. We also tested, whether the order of the 2 shRNA expression cassettes influences the extent of silencing. No significant differences were observed for the 2 plasmids, SiDEx-shDAF2-shRdRP5 and SiDEx-shRdRP5-shDAF2 (Fig. 2c and d), and thus the former plasmid was used for all subsequent experiments.
Downregulation of endogenously expressed DAF
The success of RNAi as a new antiviral strategy depends on efficient delivery systems, which can maintain long-term shRNA expression to achieve durable gene silencing. Recombinant AAV vectors are promising vehicles for efficient gene transfer with a low risk of toxic side effects. The recently developed scAAV vectors allow rapid and strong expression of a transgene in transduced cells. The cell tropism of the vector is determined by the serotype of the AAV vector (Daya and Berns, 2008). Initial experiments were carried out to investigate whether an scAAV vector with the genome and capsid protein from serotype 2 is suitable to transduce RD and HeLa cells, which were used for further experiments. To this end, an AAV vector expressing GFP as a reporter was employed. Analysis by fluorescence microscopy revealed that virtually 100% of the RD and HeLa cells, respectively, were transduced by the AAV vector.
On the basis of these results, scAAV2 vectors were produced that harbor expression cassettes for shDAF2, shRdRP5, or a combination of both. In the shuttle plasmid, the shRNA expression cassettes are flanked by 2 inverted terminal repeats that are essential for packaging of scAAVs (Fig. 3a). As a first test, we investigated whether the scAAV2.2 vectors with the expression cassette shDAF2 or the double-expression cassette SiDEx-shDAF2-shRdRP5 were capable of silencing DAF in HeLa cells. The cells were transduced with either 1000 or 5000 vg/c. As a control, an scAAV vector was used that expressed a nontarget shRNA (AAV2.2-C). As shown in Fig. 3b, both vectors were capable of inhibiting expression of DAF after transduction with 5000 vg/c, while the control vector did not affect DAF expression.

AAV vectors expressing shRNAs against RdRP and DAF. (
Inhibition of EV-30 by SiDEx-scAAVs
Finally, the shRNA-expressing scAAV2 vectors were employed to protect RD cells from the cytopathic effect, which is caused by EV-30 (Johnston and Siegel, 1990). To achieve an optimal antiviral effect, the 2 cassettes expressing shRNAs against the RdRP of EV-30 (shRdRP5) and DAF (shDAF2) were combined. For analysis of the antiviral activity, cell viability assays were performed. RD cells were pretreated with either 1000 or 5000 vg/c of shRNA-expressing scAAV2 vectors or with 5000 vg/c of the control vector. Three days post-transduction, cells were infected with EV-30 at a multiplicity of infection of 0.1 (ie, 1 virus particle per 10 seeded cells). As shown in Fig. 4, pretreatment of the cells with either scAAV2-shRdRP5 or the double-expression vector scAAV2-SiDEx-shDAF2-shRdRP5 resulted in a potent inhibition of EV-30 48 hours postinfection.
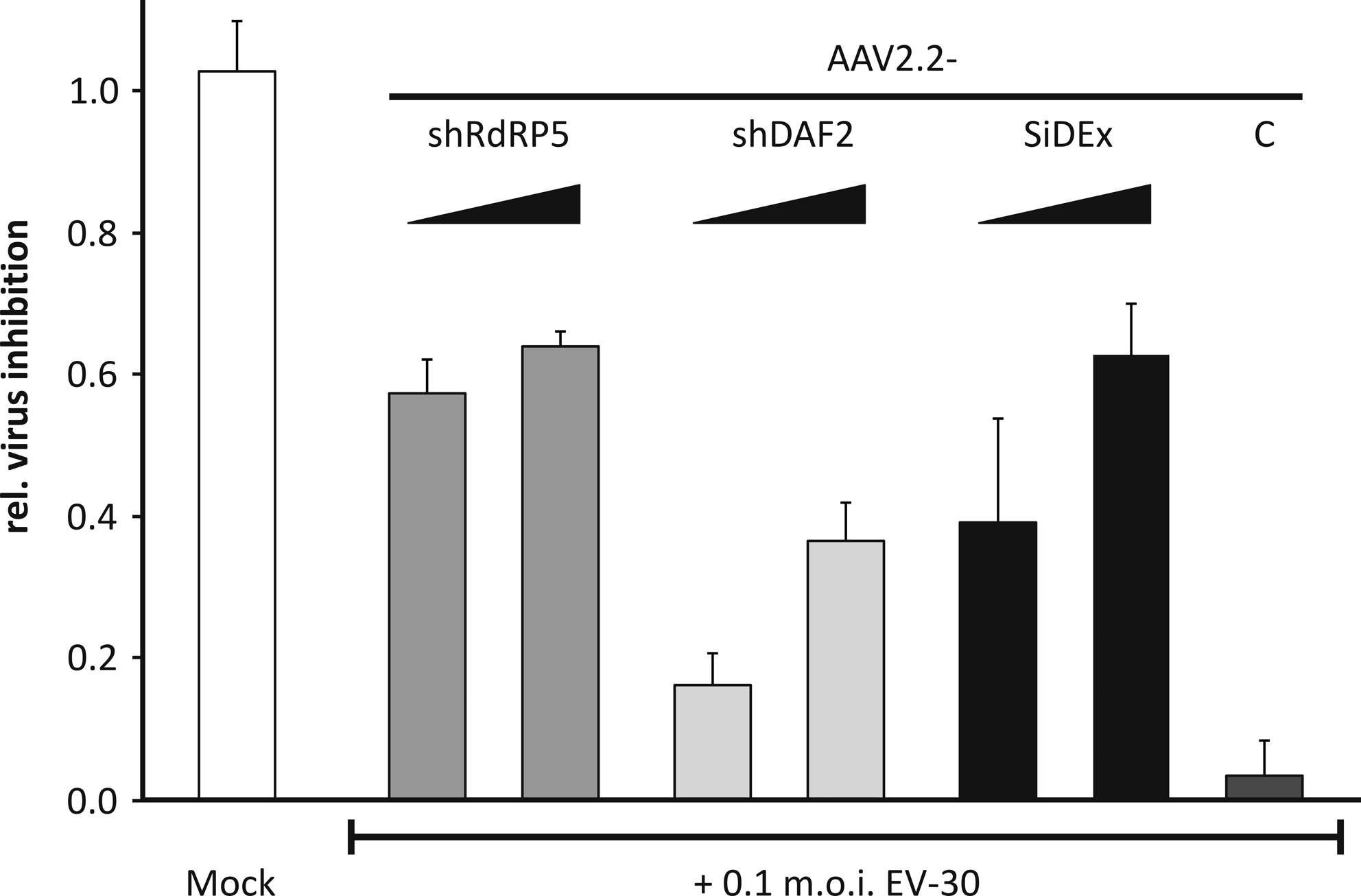
Antiviral activity of scAAV2 vectors expressing shRNAs against RdRP and DAF. RD cells were transduced with either 1000 or 5000 vg/c and subsequently infected with 0.1 m.o.i. of EV-30. Cell viability was measured 48 hours postinfection. Values were normalized to cells that were transduced with an empty scAAV2 vector, but not infected with the virus. Averages and standard deviations of 3 experiments are shown. C, cells transduced with 5000 vg/c of the scAAV2 vector expressing the control shRNA; Mock, untransduced and uninfected cells; m.o.i., multiplicity of infection.
Discussion
In recent years, RNAi has proven to be a powerful approach against viruses. One of the main advantages of RNAi as compared to the use of small molecules is the opportunity to rapidly adjust the strategy to new virus variants or even to previously unknown types of viruses. However, the design of highly efficient siRNAs against viruses still remains a challenging task, while active siRNAs are available for almost any given cellular target. We recently provided a systematic approach to design siRNAs with high potency against a virus-specific target site (Rothe et al., 2009). The applicability of an siRNA as an antiviral therapeutic is still limited by poor delivery into target cells and rapid enrichment of resistant mutants. In the present study, we developed a rapid procedure to build up a vector for the delivery of multiple shRNA expression cassettes. As a first step, several shRNA expression cassettes were generated by simultaneous cloning. Interestingly, some of the siRNAs with high antiviral activity (Rothe et al., 2009) had a diminished efficacy as the corresponding shRNA. The reason for this observation is not fully understood. It might either be due to a reduced expression level of the shRNA or to incorrect folding and maturation of the expressed shRNA. In a recent publication, Schopman et al. (2010) made a direct siRNA–shRNA comparison, which, in line with our findings, revealed that no tight relationship between the activity of siRNA–shRNA pairs exists. In further experiments, they showed that the loop length and sequence of the shRNA have great impact on the silencing activity. By optimizing the loop they were able to drastically enhance the efficiency of weak shRNA inhibitors.
The shRNA expression cassettes generated by the parallel cloning procedure were equipped with a restriction site for MfeI. Cleavage with this restriction enzyme generates ends that are compatible with EcoRI and thus allow the combination of 2 or more shRNA expression cassettes in a building block manner. The combination of shRNAs against a virus and/or its receptor on the host cell will help to minimize the risk of viral escape. A similar cloning strategy has recently been independently proposed for the generation of a vector that was intended to silence 2 endogenously expressed genes simultaneously (Xu et al., 2009).
The multiple shRNA expression cassettes obtained by the building block principle can subsequently be transferred into shuttle plasmids for the production of AAV vectors. For the applications used in the present study, standard vectors based on serotype 2 were found to be suitable to transduce virtually all cells in RD and HeLa cultures, respectively.
In a previous study, we have demonstrated that DAF plays an important role either as attachment factor or as a receptor for EV-30 (Rothe et al., 2009). We therefore tested the ability of the AAV vectors expressing shDAF2 to silence the endogenously expressed gene in HeLa cells. As can be seen in Fig. 3, AAV2.2-shDAF2 silences DAF in a dose-dependent manner. The SiDEx vector seems to be slightly less active at the lower vector dosis of 1000 vg/c, which might be due to a competition of the 2 shRNAs for components of the RNAi machinery. At the higher dosis of 5000 vg/c, expression of DAF is also inhibited virtually to completion with the shRNA double-expression vector. It should be noted that the aim of using a SiDEx vector is not to increase the potency, but rather to inhibit 2 targets simultaneously, for example, for preventing viral escape.
Finally, we tested the efficacy of either the individual shRNA expression cassettes shDAF2 and shRdRP5, respectively, or their combination to inhibit EV-30 in cell viability assays. While shRdRP5 efficiently blocked the virus, knockdown of DAF resulted in partial inhibition of the virus only. This finding is in line with our previous data obtained with siRNAs (Rothe et al., 2009) and might be due to the fact that residual DAF on the cell surface after RNAi treatment might be sufficient to allow a significant fraction of the virus to enter the cell. Importantly, the double-expression vector, scAAV2-SiDEx-shDAF2-shRdRP5, led to strong inhibition of the virus at 5000 vg/c. In contrast to the vector expressing only the shRNA against the viral RNA, the double-expression system can be expected to prevent the emergence of escape mutants, which were shown to rapidly occur after targeting enteroviruses with a single siRNA (Merl and Wessely, 2007) and thus maintain virus inhibition for a prolonged period.
Interestingly, the combination of shRNAs did not result in an additive inhibitory effect. This finding is in contrast to other reports, in which a combination of 2 siRNAs (targeting CXCR4) resulted in enhanced gene silencing compared with each single siRNA (Ji et al., 2003). In another publication, the combined expression of shRNAs resulted in a much stronger inhibition of HIV production than a single shRNA (ter Brake et al., 2006). Other groups, however, did not observe such effects. For example, Ahn et al. (2005) did not find additive antiviral effects of 2 shRNAs against CVB-3, and Koller et al. (2006) even reported that 2 siRNAs may compete for RNA-induced silencing complex (RISC) binding and thereby reduce each other's activity. We carried out additional experiments and found the inhibitory activity of an antiviral siRNA to decrease upon addition of a second (unspecific) siRNA (data not shown). It is, however, possible that an additive effect might have been missed in the concentration range used in the latter experiments.
In the present study, we generated shRNA double-expression cassettes and confirmed the silencing activity of both shRNAs. Further, we have shown that the efficiency is independent of the order of the 2 shRNA expression cassettes both of which contain a U6 promoter. For enteroviruses, the combination of 2 shRNAs was reported to be sufficient for a complete cure of a cell culture persistently infected with poliovirus (Saulnier et al., 2006) and 2 siRNAs were found to drastically reduce the risk of viral escape for CVB-3, while a cocktail of 3 siRNAs was found to be even more efficient (Merl and Wessely, 2007). For other viruses such as HIV, a combination of up to 4 shRNA expression cassettes was suggested (ter Brake et al., 2008). The number of shRNAs to be combined to minimize the risk of viral escape will thus have to be determined for each specific case. It is therefore important to note that recombinant scAAV vectors can harbor transgenic material of up to 2.4 kb (Daya and Berns, 2008). Thus, up to 6 shRNA expression cassettes, each being ∼400 bp long, can theoretically be combined in AAV vectors. In our experiments presented here, we have not observed any recombination events during production of the AAV vectors, nor have we detected signs of interference between the 2 U6 promoters. For the combination of even more shRNA expression cassettes, however, it was shown that the combination of different polymerase III promoters (eg, the H1, 7SK, or U1 promoter in addition to the U6 promoter) might be advantageous to prevent recombination (ter Brake et al., 2008).
Conclusions
The parallel cloning procedure described here is a simple approach to generate a pool of shRNA expression vectors that can be tested easily in GFP reporter assays. The insertion of an additional MfeI site provides the option to generate double or multiple shRNA expression vectors for simultaneous knockdown of viral or host cell targets. Active expression cassettes can be transferred into AAV vectors that allow efficient delivery into target cells and long-term expression of shRNAs for durable gene silencing or virus inhibition.
Footnotes
Acknowledgments
The authors thank S. Niedrig for excellent technical assistance, D. Werk for fruitful discussions, and E. Wade for critical reading of the article. H.Z. is grateful for support from Otto-Kuhn-Stiftung im Stifterverband für die Deutsche Wissenschaft. Financial support from the Deutsche Forschungsgemeinschaft (SFB TR 19, TP C1, FE 785/3-1 and KU 1436/6-1) is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.