
Abstract
Abstract
Phosphorylation dysregulation has been implicated in various diseases including cancer. The phosphorylation change of proteins in secretome may be a novel source for the discovery of biomarkers and drug targets. In this study, the phosphoproteins in cancer secretome (phosphosecretome) were globally analyzed for the first time by phosphoproteomics. One hundred forty-two phosphorylation sites on 62 unique phosphopeptides representing 49 nonredundant proteins were identified, several of which are known as secreted proteins involved in carcinogenesis, invasion, and metastasis. Most of them were first found as secreted proteins with no previously known function. Protein sublocation analysis showed that 33 proteins were found to be sectreted as phosphoproteins, in which 27 (81.81%) were secreted by a nonclassic, ER/Golgi-independent pathway, suggestting that the phosphorylation modification of these proteins might play an important role in their nonconventional secretion processes. Their protein kinases and regulatory phosphosites involved in the secretion regulation of these phosphoproteins, such as stanniocalcin 2, annexin A2, and HSP90 alphạ, were first identified. The phosphosecretome data enriched the secretome database and phosphoproteome database, and will help us to discover cancer biomarkers and drug targets, illustrating the mystery of the nonclassic protein secretion pathway.
Introduction
Protein secretion pathways include a classic, endoplasmic reticulum (ER)/Golgi-dependent secretory pathway and a nonclassic, ER/Golgi-independent pathway. In the classic secretory pathway, soluble secretory proteins typically contain N-terminal secretion signal sequences that guide proteins into the ER lumen via the signal-recognition particle. The secretion signal sequences are then cleaved and the mature proteins are transported through the Golgi to the extracellular microenvironment via an exocytosis mechanism (Mellman and Warren, 2000). However, a novel, unconventional protein secretion pathway from eukaryotic cells was reported in 1990. In this report, interleukin 1β̣ and galectin-1 were found to be released from cells into the extracellular microenvironment in the absence of the classic ER/Golgi-dependent secretion system (Cooper and Barondes, 1990; Rubartelli et al., 1990). The nonclassic protein secretion pathway was devoid of ER/Golgi-dependent postmodifications such as N-glycosylation. Although the molecular mechanism mediating the ER/Golgi-independent secreteion process remains poorly known, nonclassic protein secretion was found to be dependent onto both energy and temperature manner (Nickel, 2003) and also be regulated by various factors, including protein kinases (e.g., p38, CK2) (Jellinek et al., 2000; Santhamma et al., 2004), the NFκB signaling pathway (Wakisaka et al., 2002), and postmodifications (e.g., phosphorylation) (Deora et al., 2004; Maizel et al., 2002; Mougous et al., 2007).
Protein phosphorylation plays a crucial role in controlling various biological responses, such as cell cycle, cell growth, differentiation, invasion and metastasis, and apoptosis. Phosphorylation dysregulation has been implicated in various diseases including cancer. Studies showed that some proteins were secreted into the extracellular microenvironment as phosphoproteins to exert their biological functions such as heat-shock protein 90 alpha (HSP90α) (Wang et al., 2009), HSP70 (Mambula and Calderwood, 2006), HSP27 (Duran et al., 2007), chicked Engrailed 2 (cEN2) (Maizel et al., 2002), high-mobility group box 1 protein (HMGB1) (Oh et al., 2009), bone morphogenetic protein 15 (BMP15) (McMahon et al., 2008), growth and differentiation factor 9 (GDF9) (McMahon et al., 2008), fibroblast growth factor-2 (FGF2) (Chen et al., 2007), Apin (Moffatt et al., 2008), and peptide hormones including stanniocalcin 1 (STC1), STC2, adrenocorticotropin, human chorionic gonadotropin, atrial natriuretic peptide, progastrin, growth hormone, prolactin, and so on (Jellinek et al., 2000). These reports showed that these phosphoproteins in secrotome played an important role in inflammation, folliculogenesis, carcinogenesis, invasion, and metastasis. The phosphorylation modification is essential for their bioactivity. For example, the dephosphorylated forms of rhBMP-15 and rhGDF-9 can abolish the bioactivity of rhBMP-15, rhGDF-9. Large-scale phosphoproteomic analyses in some cancer cells (Kim et al., 2005; Rikova et al., 2007), mouse liver (Villen et al., 2007), human pituitary (Beranova-Giorgianni et al., 2006), cerebrospinal fluid (Bahl et al., 2008), and some organelles such as nucleus (Beausoleil et al., 2004), spindle body (Malik et al., 2009), and mitochondria (Boja et al., 2009), have been reported; however, the phosphoproteins in secretome (phosphoscretome) have not been globally characterized.
Here, we optimized a sample preparation to analyze the phosphoproteome in conditioned culture medium from cultured gastric cancer cells. In total, 142 phosphorylation sites on 62 unique phosphopeptides representing 49 nonredundant proteins were identified. Protein sublocation analysis showed that 33 phosphoproteins were the sectreted proteins, in which 27 (81.81%) were secreted by the nonclassic, ER/Golgi-independent pathway. The phosphosecretome data may provide another source for biomarker and drug-target discovery.
Materials and Methods
Cell culture and sample preparation
Gastric cancer SGC-7901 cells were grown in the RPMI1640 medium with 10% new-born calf serum plus 10 mM HEPES buffer. The conditioned medium was prepared as previously described with minor modifications (Gronborg et al., 2006). For Trypan blue staining and Western blot analysis, the cells were grown to 90% confluence, subsequently washed six times using serum-free RPMI1640 medium, and further incubated for 4, 6, 8, 10, and 12 h in serum-free medium (SFM) in 5% CO2 at 37°C, respectively. For phosphopeptide enrichment and identification, approximately 5.6 × 107 cells were incubated for 6 h in the SFM. The conditioned meduim containing the secreted proteins was harvested, centrifuged for 5 min at 2,000 rpm, filtered using a 0.22-μm filter, and subsequently concentrated, desalted, and washed three times with 3 mL of 25 mM NH4HCO3 by using a 5,000-Da molecular mass cutoff centrifugal filter device (Millipore, Bedford, MA), and finally exchanged into about 100 μL solution.
Trypan blue staining
The 1.0 × 106 cells cultured in conditioned medium for 0, 4, 6, 8, 10, and 12 h were suspended, and the 100 μL of cell suspension was gently mixed with equal volume of 0.4% trypan blue for 5 min at room temperature. A total of 10 μL of the stained cells was placed in a Hemocytometer, the number of viable (unstained) and dead (stained) cells were counted and photographed. The conditioned medium from 1.0 × 106 cells for different times as above were also stained with 0.4% trypan blue and counted under a microscope.
Western blot analysis
All the secreted proteins from the 4.0 × 106 cells cultured in SFM for 6 h were separated by 10% SDS-PAGE and then electroblotted onto polyvinylidene fluoride (PVDF) membranes. The proteins were incubated with antiserine phosphorylation antibodies (Invitogen, Carlsbad, CA, USA) and antityrosine phosphorylation p-Tyr-100 antibody (Cell Signaling, Danvers, MA, USA) at 4°C overnight, respectively, followed by incubation with corresponding secondary antibodies at room temperature for 1 h. The phosphorylation levels of proteins were detected using the SuperSignal chemiluminescence system (Pierce, Rockford, IL, USA), followed by exposure to autoradiographic film at darkroom.
Protein digestion
The secreted proteins were digested as previously described (Yan et al., 2006). Briefly, the proteins were reduced in 10 mM DTT at 37°C and alkylated with 20 mM iodoacetamide at room temperature, followed by precipitation in ice-cold buffer (50% acetone, 50% ethanol, 0.1% HAc). The precipitated proteins were dissolved by 50 mM NH4HCO3 and digested with trypsin (1:25, w/w) overnight at 37°C. The digested solutions were evaporated to about 20 μL in a SpeedVac centrifuge for phosphopeptide enrichment analysis.
Phosphopeptide enrichment using TiO2
Phosphopeptide enrichment was performed as described in the Proteoextract phosphopeptide enrichment TiO2 kit user manual (Calbiochem, LaJolla, CA, USA). In brief, trypsin-digested peptides were diluted with TiO2 Phosphobind Buffer containing 2,5-dihydroxybenzoic acid (DHB). The diluted peptide mixtures were added to the TiO2 Phosphobind resin, and incubated with gentle agitation for 10 min at room temperature, followed by washing TiO2 Phosphobind resin with Wash Buffer. The phosphopeptides were eluted from the TiO2 Phosphobind resin by elution buffer and 300 mM NH4OH/50% CH3CN, respectively. The eluted supernatant was pooled and evaporated to dry in SpeedVac centrifuge for nano-SCX-LC-MS/MS analysis.
Nano-SCX-LC-MS/MS analysis
Strong cation exchange and liquid chromatography coupled with LTQ-Orbitrap mass spectrometry (Thermo Electron, Bremen, Germany) was used to analyze the enriched phosphopeptides as previously described with minor modification (Pan et al., 2008). In brief, the phosphopeptides loaded on a SCX column were eluted by NH4Cl with nine different concentrations (0, 5, 10, 40, 80, 200, 320, and 500 mM, and 1 M). Subsequently, the eluted solution was entered into a C18 reverse phase column, which was further eluted with a 0–40% gradient buffer solution (Buffer A, 0.1% formic acid, and 5% AcN; Buffer B, 0.1% formic acid, and 95% AcN). The eluted phosphopeptides were then online analyzed by TOP 5 method in LTQ-Orbitrap mass spectrometer with capillary temperature of 200°C and spray voltage of 1.80 kV as previously described with minor modification (Pan et al., 2008). A full MS scan with mass range of 350–1,800 m/z was carried out in the Orbitrap, and followed by 5 MS2 scans in the LTQ. If an ion has a neutral loss of 98.00 (loss of a phosphate group with a charge state), 58.00 (loss of a phosphate group and H2O with two charge states), 49.00 (loss of a phosphate group with two charge states), 38.67 (loss of a phosphate group and H2O with three charge states), or 32.67Da (loss of a phosphate group and H2O with three charge states), and is one of the five most intense ions in MS2 scans, MS3 scans were further carried out in the LTQ. Conditions with 35% normalized collision energy, activation q of 0.25 and activation time of 30 ms were applied for both MS2 and MS3 acquisitions.
Phosphopeptide identification and phosphosite validation
The derived peak lists extracted by DTASuperCharge V1.31 (SourceForge) were searched against a real and false IPI human database (V3.56) containing 153,078 human protein entries by Mascot 2.2.04 search engine (Matrix Science, London, UK). The search parameters were as follows: precursor ion mass tolerance and fragment ion mass tolerance were 10 ppm and 0.5 Da, respectively; the maximum of the missed cleavages were 2; Carbamidomethylation was as fixed modification, Oxidation (M), Phospho (ST), and Phospho (Y) were as variable modifications.
To evaluate the false-positive rate (FPR), all spectra and all sequence assignments produced by Mascot 2.2.04 were further processed and validated with the MSQuant 1.5 software for posttranslational modification (PTM) score analysis as previously described (Olsen et al., 2006). The estimated FPR based on the decoy database search was less than 2% when peptide score threshold and PTM score threshold were 19 and 14, respectively. All spectra of these phosphopeptides were further confirmed by manual interpretation of MS/MS ion spectra using the criteria as previously described (Macek et al., 2007; Nichols and White, 2009).
For the phosphopeptides with multiple potential phosphorylation sites, the probabilities for phosphorylation at each site were evaluated based on PTM scores as previously described (Olsen et al., 2006). Phosphorylation sites with localization probability >0.75 were reported as Class I phosphosites, the probability between 0.75 and 0.25 as Class II sites. Phosphorylation sites with localization probability <0.25 were discarded.
Secretion pathways predicted by bioinformatics analysis
The sequence of each protein was submitted to a signalP and targetP program to search for a secretion signal peptide within the protein sequence when the parameters were used as default settings, respectively. The predicted results of both the signalP and targetP program were same. The sequence of each protein was submitted to the secretomeP program to analyze the nonclassical and leaderless secretion of proteins when the parameters were as default settings.
Results and Discussion
Evaluation of secretome preparation
The methods frequently used for collecting cancer secretome are to obtain the released proteins or peptides from the SFM in vitro cancer cell culture (Chevallet et al., 2007; Kratchmarova et al., 2002). However, the cells cultured in SFM tend to autolyse and liberate cytosolic proteins. Therefore, it is necessary to optimize the incubation time to minimize the cytosolic protein contamination. In our experiments, we used two methods, two cytosolic proteins, α-tubulin and β-actin, and trypan blue staining, to monitor cell autolysis (Gronborg et al., 2006; Xue et al., 2008). We found that the level of α-tubulin increased in the conditional medium in a time-dependent manner after 6-h incubation, but the level of β-actin could not be detected in the conditioned medium from 4 to 12 h incubation (Fig. 1). Traditionally trypan blue staining was used to monitor the cell death and autolysis (Gronborg et al., 2006). Surprisingly, we here found that no dead cells were detected after 4, 6, 8, 10, and 12 h of starvation and under normal culture condition by Trypan blue staining, although the cytosolic protein α-tubulin could be detected in the conditioned medium, suggesting that the detection of cytosolic proteins are more sensitive and reliable than Trypan blue staining in monitoring the cell death and autolysis in serum-free culture condition. However, we used Trypan blue to stain the conditioned-medium of 1 × 106 cells after a 6-h incubation and found that cells or cell debris floated in conditioned medium were stianed by Trypan blue. Based on these observations, we chose 6 h as the suitable incubation time in which dead cells and cytosolic protein contamination can be minimized. The results also showed that it was more suitable and sensitive that Trypan blue staining was used for monitoring the dead cells and cell debris floating in the supernatant, instead of the serum-free cultured cells.
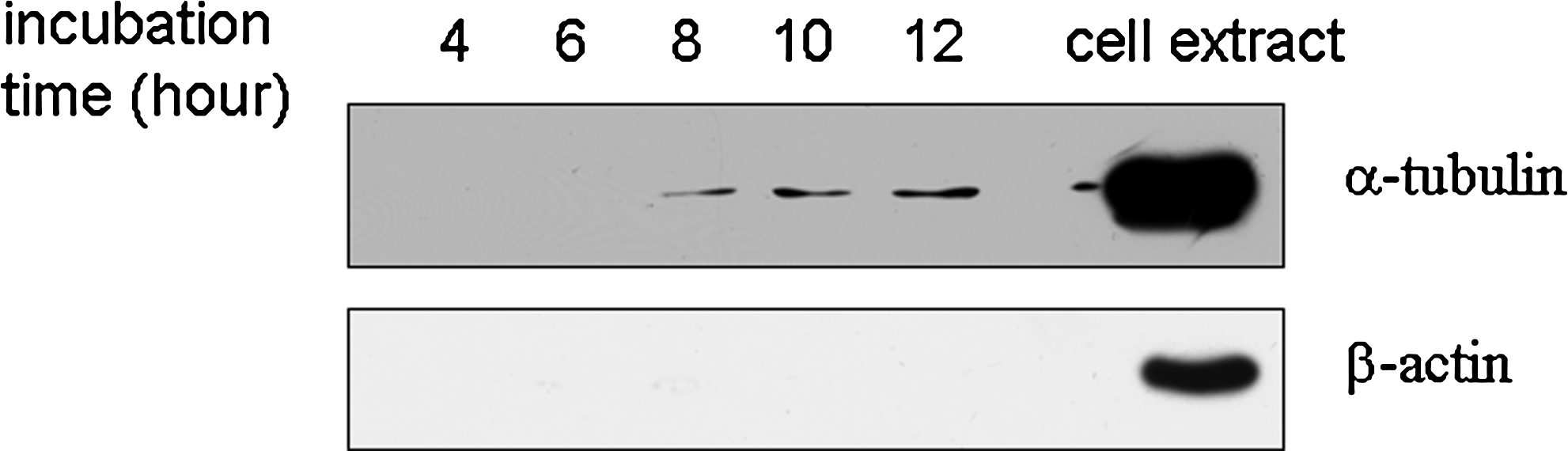
The amount of representative cytosolic proteins in the different incubation time in conditioned medium. All the conditioned medium from 1.0 × 106 cells with the different incubation time (4, 6, 8, 10, and 12 h) was collected, desalted, and concentrated, respectively. The cytosolic protein α-tubulin and β-actin in all the concentrated conditioned medium from 1.0 × 106 cells were detected by Western blotting.
Phosphorylation in secretome
Previous studies have shown that many proteins could secret into the extracellular microenvironment as phosphoproteins by an unconventional secretion pathway (Danielsen et al., 2003; Deora et al., 2004). To determine whether other phosphoproteins also existed in the cancer secretome, we analyzed the phosphorylation profile of gastric cancer secreteome by Western blotting using antiserine and -tyrosine phosphorylation antibodies, respectively. Figure 2 showed that some secreted proteins in cancer secretome were phosphorylated, and these phosphoproteins in secreteome were different from those in cells by comparing them with the phosphorylation profile in cell extracts, suggestting that other novel phosphoproteins existed in cancer secreteome.
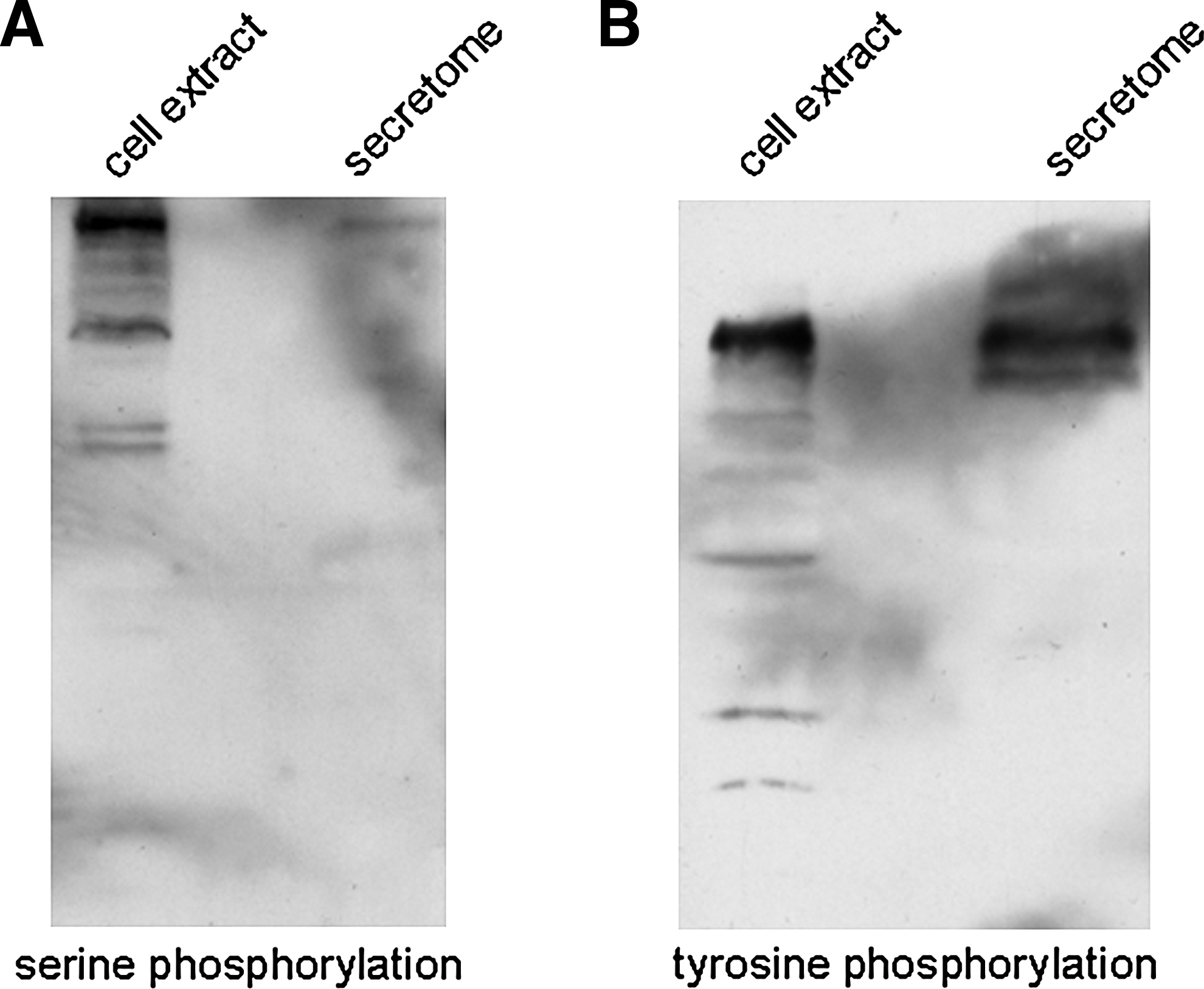
Some proteins in conditioned medium were found to be serine- and tyrosine-phosphorylated by Western blotting analysis. Approximately 4.0 × 106 cells were incubated for 6 h in serum-free medium, and all the conditioned mediums were collected, desalted, and concentrated. The phosphorylation of secreted proteins in concentrated conditioned medium from 4.0 × 106 cells were detected by Western blotting using antiserine phosphorylation (
Identification of phosphorylated proteins and sites in secretome
To identify phosphoproteins in secretome, the supernatant from 5.6 × 107 gastric cancer cells was collected, concentrated, desalted, digested with trypsin, and the resulting peptides were subjected to TiO2 phophopeptide enrichment followed by online nano-SCX-LC-MS/MS analysis in a LTQ-Orbitrap mass spectrometer. The MS/MS spectra were searched against the human IPI protein database (V3.56) containing sequences in forward and reverse directions by using a Mascot search engine for the phosphorylation assignment.
A total of 142 phosphorylation sites, mapping to 62 unique peptides and 49 different proteins, were identified at an FPR of less than 2%. To precisely assign the ambiguous phosphosites within a peptide, the probabilities of phosphorylation at each site were calculated based on PTM score as previously described (Olsen et al., 2006). Among the 142 phosphorylation sites, we could localize 69 (48.6%) phosphosites with high confidence as a Class I phosphorylation site. The entire dataset containing the phosphorylation proteins and sites identified in the study is summarized in Supplementary Table S1. Figure 3 shows the representative MS/MS spectra for phosphosite-containing peptides in the detection; all other MS/MS spectra are available via the hyperlinks provided in Supplementary Table S1.
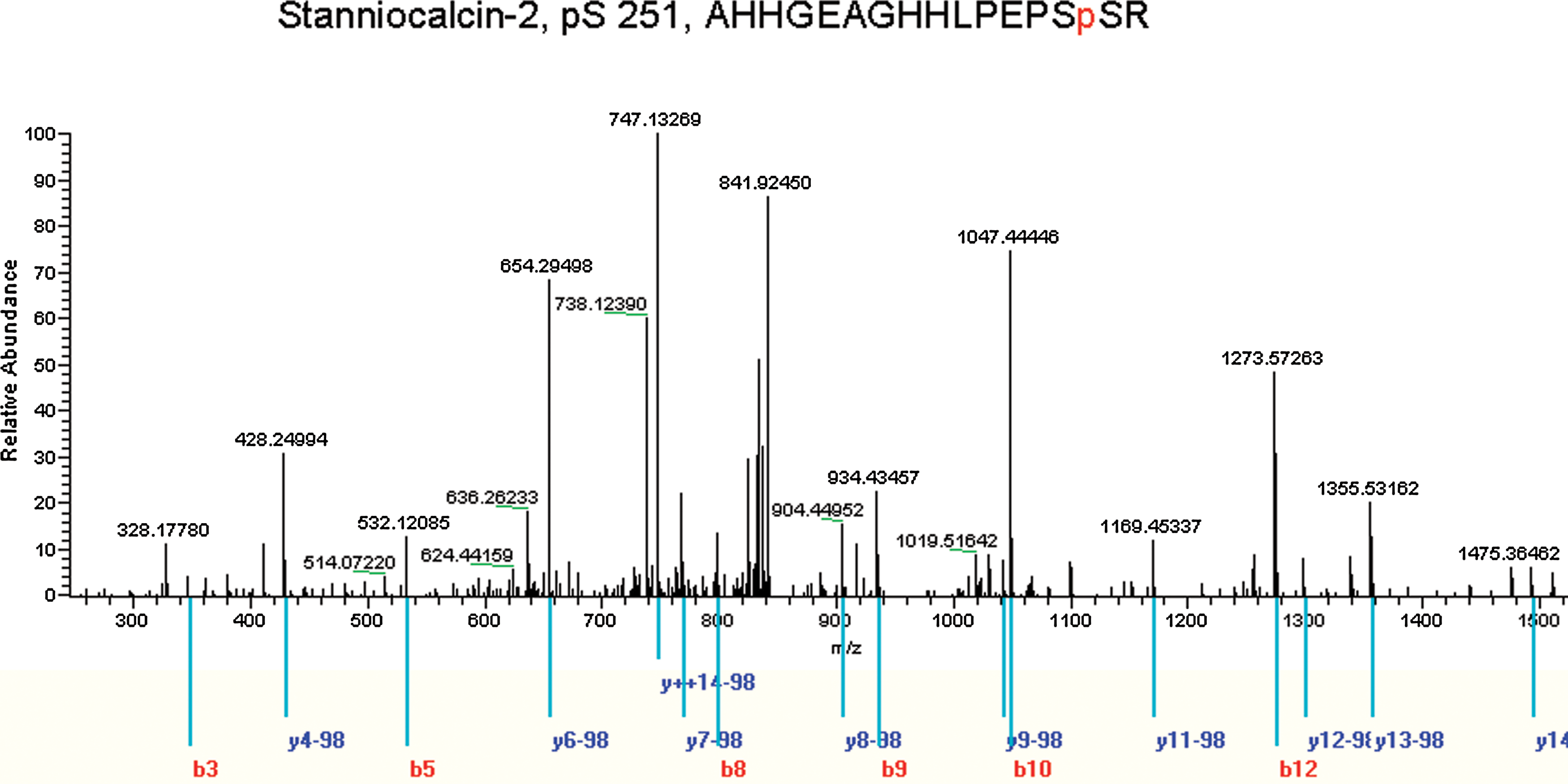
Representative MS/MS spectra for phosphopeptides. MS/MS spectra of the phosphopeptide AHHGEAGHHLPEPSpSR, which contians the phosphoserine at S251 of STC2. In the MS/MS analysis, b and y ions of various peptides were detected, and the localization of the phosphosites were determined by PTM scores in MSQuant software analysis as described previously (Olsen et al., 2006). The matched b and y ions for a given phosphopeptide sequence are colored red and blue, respectively. More information about peptide could be found in Supplementary Table S1.
Secretion pathways of these proteins
To determine whether our identified phosphoproteins could be secreted, each phosphoprotein was first searched against the Uniprot database (release in March 3, 2009) and analyzed with Gene Ontology (GO). The sublocation annotation analysis showed that 18 of our identified phosphoproteins have been reported as secreted proteins. Among them, annexin A2, SEPT2, SEPT9, BAT3, FLOT2, NSFL1C, and HSP90α have been reported to be secreted by nonclassic secretion pathway (Danielsen et al., 2003; Deora et al., 2004), and GOLM1, COPB2, and SPTBN1 are secreted by a classic, ER/Golgi-dependent pathway because they are Golgi membrane-related proteins. The secretion pathway of the remaining eight proteins is still not known; they might be secreted by a nonclassic pathway due to the absence of an N-terminal secretion signal peptide. We further used signalP, targetP, and secretomeP programs to predict whether these unannotated proteins could be secreted or not, and their secretory pathway (Bendtsen et al., 2004). Among 49 identified phosphoproteins, Bioinformatics analysis showed that 19 could be secreted in a nonclassic secretory pathway; only three were secreted via a classic pathway because they contained an N-terminal signal peptide. Taken together, as shown in Table 1, a total of 33 phosphoproteins could be secreted to extracellular space among these 49 proteins.
More information about these proteins could be found in the Supplementary Table S1 by Accession No.
The results were predicted by the secretomeP program.
The results were predicted by both the signalP and targetP programs.
The data were annonated by GO or reported by the literatures.
These proteins was related with Golgi membrane.
Interestingly, most (81.81%) of the 33 secreted proteins were secreted in a nonclassic secretory pathway. Although the previous reports have reviewed the likely nonclassic secretion mechanism of some proteins, such as FGF-1, FGF-2, galectin, IL-1, HMGB1, and viral proteins (e.g., HIV-tat and VP22) (Nickel, 2003), the accurate mechanisms were still poorly understood. Some studies have shown that protein phosphorylation modification palyed an important role in the nonclassic, ER/Golgi-independent secretory pathway (Deora et al., 2004; Maizel et al., 2002; Mougous et al., 2007). Among our identified secreted phosphoproteins, the nonclassic secretion of annexin A2, stanniocalcin-2 (STC2), HSP90α, and PCSK9 have shown to be regulated by their phosphorylation modification (Danielsen et al., 2003; Deora et al., 2004; Dewpura et al., 2008; Jellinek et al., 2000). For example, stanniocalcin 2 (STC2) was shown to be secreted into the extracellular environment as phosphoprotein, to promote cancer invasion and metastasis (Jellinek et al., 2000; Volland et al., 2009). In the microorganism, some studies also found that phosphorylation modification was invovled in the regulation of protein secretion. For example, Mougous et al. (2007) found that threonine phosphorylation regulated the Hcp1 protein secretion in Pseudomonas aeruginosa via an eukaryotic-like threonine kinase–phosphatase mechanism. These reports suggested that phosphorylation modification might play a critical role in their nonclassic secretory processes of our identified proteins, given that our identified secreted proteins were phosphorylated in secretome.
Among the 49 identified phosphoproteins, 16 were not annotated or predicted as secreted proteins. The likely reasons include (1) the secretomeP program cannot fully predict all proteins that were secreted in the nonclassic, ER/Golgi-independent pathway, because the nonconventional secretory mechanism is still poorly understood; (2) these phosphoproteins were in lower abundance than other nonphosphorylated secreted proteins, and thus were not detected by traditional secretome analysis. Although they were not annonated or predicted as secreted proteins, some of them have family members to be shown to secrete from tumor cells into the extracellular microenvironment. For example, HSP70, HSP27, and HSP90α α̣in the HSP family, which is an intracellular molecular chaperone and normally localizes in the cytoplsam and nucleus, can be secreted from tumor cells in the nonclassical secretory pathway and function as an intercellular signaling ligand (Mambula and Calderwood, 2006), suggesting that our identified other member of the HSP family, HSP90β, may be also released from cancer cells into the microenvironment in the nonclassical secretory pathway.
Surprisely, we found that some transcriptical regulators, such as Bcl-2-associated transcription factor 1 (BCLAF1), tumor suppressor p53-binding protein 1 (TP53BP1), and HIVEP2, can be secreted from gastric cancer cells as phosphoprotein. This corresponds to the observation by Maizel et al. (2002), in which transcriptional factor chicked Engrailed 2 (cEN2) homeoprotein was found to be secreted by the nonconventional secretory pathway, and the secretion could be inhibited by the phosphorylation of a serine-rich domain within cEN2 regulated by protein kinase CK2. This showed that some transcriptical regulators might be exported into the extracellular microenvironment under some conditions such as the phosphorylation regulation. Therefore, it is not surprising that our identified transcriptical regulators existed in secretome, and their phosphorylation may be associated with their nonclassic secretion.
Characterization of phosphorylation of these secreted phosphoproteins
To reveal novel phosphorylation proteins and sites, peptide sequences for phosphorylation were adjusted to ± 6 aa from the central position and then were matched to the Phosphosite database, the most comprehensive database of phosphorylation data. Supplementary Table S2 lists the matching results. One hundred eight (76.05%) of our identified phosphorylation sites have previously been observed in other studies; 24% phosphorylation sites and 8 phosphorylation proteins detected in this work have not been described in the in vivo experiments in the Phosphosite database. These novel phosphorylation sites and proteins may implicate new functions for these molecules, and their biological significance in gastric cancer will be further explored in the next step.
The phosphorylation of proteins was regulated by various protein kinases. For example, the phosphorylation of secreted stanniocalcin 2 (STC2), chicked Engrailed 2 (cEN2) was regulated by casein kinase 2 (CK2) (Jellinek et al., 2000; Maizel et al., 2002). Phosphorylation specificity of protein kinases on downstream substrates typically depends on the amino acid sequence surrounding the target phosphorylation site. To discover protein kinases that were involved in the phosphorylation regulation of these secreted phosphoproteins, the Uniprot IDs of these secreted proteins were simultaneously submitted to the Scansite program to search for specific kinases involved in the specific phosphosites identified (Obenauer et al., 2003). At the highest stringency level, Scansite prediction showed that the phosphorylation of our identified secreted proteins were mainly regulated by CK2, ERK1, and GSK3 protein kinases.
Although some proteins have been found to be secreted into the microenvironment as phosphoproteins, their phosphorylation sites and protein kinases invovled in the regulation of secretion are still unknown. Here, the phosphorylation sites of these secreted proteins were indentified by MS/MS analysis, and protein kinases with regulating these phosphosites were analyzed by the Scansite program. For example, an earlier study reported that STC2 was secreted as phosphoprotein from human fibrosarcoma cells, being phosphorylated by protein kinase CK2 (Jellinek et al., 2000). However, the phosphorylation sites invovled in the secretion regulation are unknown. In this study, we found that the Serine 250, 251, 285, 287, and 288 of STC2 in secretome were phosphorylated. Scansite analysis further showed that the STC2 phosphorylation on Serine 287, 288, and Serine 250 were regulated by protein kinase CK2 and Clk2, respectively.
In addition to its intracellular functions, Annexin A2 was found to be secreted into the ectracellular environment by nonclassic pathway, in which annexin A2 interacts with extracellular matrix proteins and specific proteases to regulate plasminogen activaction, cell migration, and cell adhension. Annexin A2 could be phosphorylated at Serine 11 and 25 by PKC, at tyrosine 23 by pp60src (Zhao et al., 2003). The inhibition of PKC and pp60src had no effect on annexin A2 secretion (Zhao et al., 2003), suggestting that the phosphorylation at Serine 11, 25, and tyrosine 23 was not associated with annexin A2 secretion. Here, we found that the Serine 30 and 145 of annexin A2 were phosphorylated in secretome, suggestting that the phosphorylation at Serine 30 and 145 was invovled in the regulation of annexin A2 secretion. Scansite analysis further showed that Serine 30 and 145 of annexin A2 was phosphorylated by protein kinase CK2. The results showed that the annexin A2 secretion was regulated by protein kinase CK2.
Conclusions
The study was first globally characterized the phosphorylation proteins in secretome. These phosphoproteins were secreted into extracellular microenvironment mainly via the nonclassic, ER/Golgi-independent pathway, and the phosphorylation modification might play an important role in the nonconventional secretory processes. The phosphorylation sites and protein kinases invovled in the regulation of secretion were first discovered in the study. We will further define the role of these secreted phosphoproteins in secretome in gastric carcinogenesis, invasion, and metastasis.
Supporting information available
Supplementary Tables S1–S2. This information is available at http://life-health.jnu.edu.cn/phospho/p-sec/Supp_Data.zip.
Footnotes
Acknowledgments
This work was partially supported by the 2007 Chang-Jiang Scholars Program, National Natural Science Foundation of China (30973393), the Fundamental Research Funds for the Central Universities (21600201), “211” Projects grant (Biotechnology & Bioengineering Drug and Biomaterial & Tissue Enigeering).
Author Disclosure Statement
The authors declared no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.