
Abstract
Abstract
One common method used for analyzing the glycoproteome is chromatography using multiple lectins that display different affinities toward oligosaccharide structures. Much has been done to determine lectin affinity using standard glycoproteins with known glycosylation; however, a knowledge of the selectivity and specificity of lectins exposed to complex mixtures of proteins is required if they are to be used as a means of studying the glycoproteome. In the present study, three lectins (Concanavalin A, Jacalin, and Wheat Germ Agglutinin) were used to fractionate glycoproteins from two different complex environments: (1) cell membranes and (2) plasma. Reproducible enrichment of glycoproteins from these samples has been shown to result from the combined use of these lectins. However, the global glycan profiles of the released N- and O-linked oligosaccharides from the glycoproteins retained by the lectins, and from those glycoproteins that did not bind, using both these complex samples, were found to be very similar. That is, although the lectins selectively and reproducibly retained some glycoproteins, other proteins with the same attached oligosaccharide structures did not bind. Some small N- and O-glycan differences were observed in the bound fractions but there was little absolute specificity toward individual oligosaccharide structures known to have high affinity to these lectins. These data indicate that lectins are useful for fractionating glycoproteins from complex mixtures, but that the overall glycoproteome is not isolated by this approach.
Introduction
Recently, mass spectrometry has become increasingly used in glycoproteomics to (1) identify the glycosylated protein and peptides, (2) determine N- and O-glycosylation site(s), and (3) analyse the structures of the attached glycan moieties (Tissot et al., 2009). With the completion of many genome and proteome databases, identification of proteins is relatively easy, fast and automated, and usually involves (1) 2DGE for protein separation, followed by MALDI-MS or (2) LC separation of proteins or peptides, followed by ESI-MS (Lim et al., 2003; Washburn et al., 2001). Identifying glycoproteins and glycopeptides by mass spectrometry in this context can be challenging due to the low level of different glycoforms of the same protein in complex mixtures, due to the attachment of structurally different glycans (microhetereogeneity) that can occupy (to varying degrees) different glycosylation sites (macroheterogeneity). Therefore, full characterization of glycoproteins from biological systems is a challenge that can be facilitated by enriching the glycoproteins from the complex mixtures of proteins. At present, the two most used methods for capturing glycoproteins are hydrazine coupling of N-linked glycoproteins and glycopeptides to a solid support (Zhang et al., 2003) and immobilized lectin chromatography that employs the affinity of these proteins for particular glycan substructures (Yang and Hancock, 2004). The disadvantage of using the hydrazine chemistry approach is that the carbohydrates are destroyed by the oxidative chemical coupling to the hydrazine leaving the glycans no longer able to be identified and characterized. The use of lectins as a glycocapture method is considered to be a more favorable approach for glycosylation analysis because the integrity of the attached glycans is maintained during the fractionation.
Lectins are proteins found in plants, animals and microorganisms and have different affinities for particular oligosaccharide structures displayed on proteins and lipids and are frequently used in cytology and histology to stain glycoproteins (Collins et al., 1982). Several lectins with different specificities have been used sequentially (Lee et al., 2009) and in parallel (Yang and Hancock, 2004) to enrich glycoproteins from complex mixtures; to identify low abundance plasma proteins (Yang and Hancock, 2004), to examine the microheterogeneity of carbohydrates associated with disease and disease progression (Hirabayashi and Kasai, 2002; Vlassara and Palace, 2002), to identify liver membrane glycoproteins (Lee et al., 2009), and to target specific glycosylation sites on proteins (Kaji et al., 2003). Although lectins have been used to isolate glycopeptides (Hirabayashi, 2004; Hirabayashi et al., 2002; Kaji et al., 2003) the focus of this article is on investigating the role of lectins as a means of enriching glycoproteins from complex samples.
Three lectins, Concanavalin A (ConA), Jacalin, and Wheat Germ Agglutinin (WGA) are commonly used in these types of experiments and have been used as a multilectin affinity chromatography (M-LAC) system for reproducibly enriching serum or plasma glycoproteins from patients with breast cancer (Yang et al., 2006), autoimmune (Plavina et al., 2007), and metabolic syndrome diseases (Dayarathna et al., 2008). Using a mixture of lectins such as the M-LAC system has been demonstrated to increase the binding of glycoproteins compared to the use of a single lectin (Ralin et al., 2008). This is presumably due to the heterogeneity of glycosylation on individual proteins that could lead to multisite attachment between the glycoprotein and the different lectins in the M-LAC system. ConA is a lectin found in the jack bean Canavalia ensiformis that is composed of identical protomers that combine to form a tetrameric complex. Each monomer is approximately 25 kDa in size and contains one saccharide binding site, which allows for four saccharides to be bound to the lectin at any given time (Becker et al., 1975; Cunningham et al., 1975; Edelman et al., 1972; Reeke et al., 1975a). ConA primarily recognizes α-mannose residues that are arranged in a trimannosyl {Manα(1,3)[Manα(1,6)]Man} configuration, which is always part of the N-glycan core structure (Goldstein, 1975; Goldstein et al., 1965a, 1965b; Reeke et al., 1975b). Con A has increased affinity for tri-mannosyl oligosaccharide structures that are elongated by α(1–2) linked mannose (or α-glucose) residues on high mannose and hybrid N-glycan structures (Brewer et al., 1986). However, tri- and tetra-antennary N-glycans and N-glycan structures that consist of a bisecting N-acetyl-D-glucosamine (GlcNAc) or a fucosylated core show reduced or no binding to ConA (Brewer et al., 1986; Cummings and Kornfeld, 1982).
Jacalin is a lectin found in seeds of jack fruit (Artocarpus integrifolia) and is a homotetrameric glycoprotein of four identical protomers consisting of a light (β) and a heavy (α) polypeptide chain of 20 and 133 amino acid residues, respectively (Young et al., 1991). Jacalin undergoes a series of co- and posttranslational processing events including signal peptide cleavage and N-linked glycosylation to produce the mature and functional tetrameric lectin. Jacalin has been acknowledged as a general lectin for O-glycans by its recognition of the ubiquitous O-linked structure galactosyl β(1–3) N-acetyl-galactosamine [Galβ(1–3)GalNAc], although it also has affinity for lactose and galactose (Bourne et al., 2002; Roque-Barreira and Campos-Neto, 1985). In addition, X-ray crystallography and surface plasmon resonance measurements have indicated that a relatively large carbohydrate-binding site enables Jacalin to also bind a number of different monosaccharides including mannose, oligomannosides, glucose, N-acetylneuraminic acid (NeuAc) and N-acetylmuramic acid (Bourne et al., 2002; Do and Lee, 1998). The affinity for these different sugars is thought to occur at the same binding site as galactose in the O-linked disaccharide and not at two distinct binding sites with different specificities (Bourne et al., 2002).
WGA is a lectin isolated from Triticum vulgaris that functions as a dimeric protein for carbohydrate binding, with each monomer having two oligosaccharide binding sites (Kronis and Carver, 1985; Nagata and Burger, 1972, 1974). The two identical WGA monomers have a combined molecular weight of approximately 35 kDa with amino acid sequence made up of mostly glycine and half-cystine amino acids, with none of the half-cystines present as cysteines (Nagata and Burger, 1972). WGA exhibits a primary affinity for GlcNAcβ(1–4)GlcNAc and is commonly used to bind sialylated complex N-glycan structures due to its secondary affinity for NeuAcα(2,3) (Nagata and Burger, 1974). Despite its recognition of some sialylated glycans, WGA has very little or no affinity for NeuAcα(2,6) and NeuAcα(2,8) sialylated glycans (Iskratsch et al., 2009) and shows no recognition of core fucosylated N-glycan structures (Cummings and Kornfeld, 1982; Iskratsch et al., 2009).
In this study, three immobilized lectins (ConA, Jacalin, and WGA) commonly used to enrich N- and O-linked glycoproteins were applied to two different biological systems (Triton X-114 phase partitioned rat liver membranes, and albumin and IgG-depleted human plasma) to examine the differential enrichment of glycoproteins and their attached glycans. The global N- and O-glycan profiles of the lectin unbound and bound glycoproteins from rat liver membrane and human plasma were compared to try to understand the basis of the glycoprotein separation by lectin affinity chromatography in two different complex biological systems. We have previously described that these three lectins can be effectively and reproducibly used for proteomic studies to isolate subsets of glycoproteins from complex mixtures (Kullolli et al., 2010). The results presented here demonstrate that lectin affinity for fractionating glycoproteins from complex samples can have limitations for glycomic analysis, which was not resolved by this approach.
Materials and Methods
Materials
Tris, sodium chloride, ammonium bicarbonate (NH4HCO3), formic acid, Triton X-114, 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), ammonium acetate (CH3COONH4), dithiothreitol (DTT), iodoacetamide (IAA), methyl-α-glucopyranoside, galactose, N-acetyl-glucosamine, urea, thiourea, tributyl phosphine (TBP), ovalbumin from Gallus gallus, glycerol, SDS, agarose bound Jacalin with protein concentration of 5 mg/mL and binding capacity of 1–2 mg human IgA/mL, and agarose bound WGA with protein concentration of 5 mg/mL and binding capacity 1–2 mg ovomucoid/mL were obtained from Sigma (St. Louis, MO, USA). α1-Protease inhibitor (A1PI) from human serum was a donation from Baxter Bioscience, Austria. Agarose bound Concanavalin A (ConA) with protein concentration of 10–16 mg/mL and binding capacity of 20–45 mg porcine thyroglobulin/mL was obtained from GE Healthcare (Uppsala, Sweden). Sequencing-grade trypsin was obtained from Promega (Madison, WI, USA). N-Glycosidase F (PNGase F, recombinant cloned from Flavobacterium meningosepticum and expressed in Escherichia coli) lyophilized in 10 mM sodium phosphate buffer, pH 7.2 was acquired from Roche Diagnostics (Basel, Switzerland). Immobilon-P polyvinylidene fluoride (PVDF) was purchased from Millipore (Bedford, MA, USA). AG50W-X8 cation-exchange resin was obtained from Bio-Rad (Hercules CA, USA).
Rat liver membrane preparation
Rat liver membrane proteins were prepared according to our previous method (Lee et al., 2009). Briefly, rat liver tissue (∼1.5 g) was homogenized in 5 mL lysis buffer (pH 7.4) containing 50 mM Tris-HCl, 0.1 M NaCl, 1 mM EDTA, and protease inhibitor cocktail (Roche Diagnostics) using an Omni TH homogenizer (Omni International Inc., Kennesaw, GA, USA) and centrifuged at 2,000 × g for 20 min at room temperature. The supernatant containing cellular proteins was removed and diluted with binding buffer (20 mM Tris-HCl, 0.1 M NaCl, 1 mM MnCl2, 1 mM MgCl2, and 1 mM CaCl2, pH 7.4) to a final volume of 40 mL and chilled on ice for 1 h. This was to ensure that the applied sample was compatible with lectin affinity chromatography. The diluted proteins were sedimented by ultracentrifugation at 120,000 × gavg for 1 h and 20 min. The membrane pellet was washed twice with 0.1 M sodium carbonate (pH 11).
Approximately 100 μL (equivalent to approximately 0.5 mg) of the rat liver membrane pellet was homogenized with 4 volumes of binding buffer containing 1% (v/v) Triton X-114 and chilled on ice with intermittent vortexing for 20 min. The samples were heated at 37°C for 20 min and phase partitioned by centrifugation at 300 × g for 5 min. The upper aqueous phase was removed and stored and phase partitioning was repeated. The combined aqueous phases were mixed with 9 volumes of ice-cold acetone to precipitate proteins and remove any detergent. Membrane proteins contained in the Triton X-114 detergent phase were precipitated with 9 volumes of ice-cold acetone. Precipitated membrane proteins were resolubilized in binding buffer containing 1% (w/v) CHAPS, heated at 96°C for 6 min, and stored at −80°C if not used immediately. Protein quantification was performed using a Bradford Assay (Sigma-Aldrich, St. Louis, MO, USA).
Lectin affinity chromatography of rat liver membranes
Lectin affinity chromatography was carried out as previously described (Lee et al., 2009) Approximately 80 μg of detergent-partitioned proteins in 0.5 mL of binding buffer containing CHAPS was mixed with 0.25 mL of agarose-bound ConA (protein concentration 10–16 mg/mL and binding capacity of 20–45 mg porcine thyroglobulin/mL), 0.5 ml agarose-bound Jacalin (protein concentration 5 mg/mL and binding capacity of 1–2 mg human IgA/mL) and 0.5 mL of agarose-bound WGA (protein concentration 5 mg/mL and binding capacity of 1–2 mg ovomucoid/mL) and incubated at 4°C overnight. Each mixture was packed in disposable gravity-flow Poly-Prep chromatography columns (Bio-Rad, Hercules, CA, USA) and the three lectin columns were washed with 10 bed volumes of binding buffer. Bound proteins were then eluted with 10 column volumes of elution buffer A [20 mM Tris-HCl, 0.1 M NaCl, 0.2 M methyl-α-D glucopyranoside (ConA), 0.5 M α-D galactose (Jacalin), or 0.5 M N-acetyl glucosamine (WGA), pH 7.4], followed by 10 bed volumes of corresponding higher concentration elution buffer B [20 mM Tris-HCl, 0.1 M NaCl, 1 M methyl-α-D glucopyranoside (ConA), 1 M α-D galactose (Jacalin), or 1 M N-acetyl glucosamine (WGA), pH 7.4]. Further elution of tightly bound proteins was performed using 10 bed volumes of the relevant elution buffer C [20 mM Tris-HCl, 0.1 M NaCl, 1.5 M methyl-α-D glucopyranoside (ConA), 1.5 M α-D galactose (Jacalin), and 1.5 M N-acetyl glucosamine (WGA), pH 7.4]. All eluted samples were desalted and concentrated using an Amicon Ultra-10 centrifugal device with 10 kDa nominal molecular weight limit (NMWL) (Millipore). Lectin eluates with high concentrations of monosaccharides were diluted with water to assist in the diafiltration and concentration of proteins.
Lectin affinity chromatography of human plasma proteins
Human plasma glycoproteins were prepared as described (Kullolli et al., 2010). Briefly, 2 mL of human plasma (from a normal female volunteer) was depleted using a protein A and polystyrene-divinylbenzene support matrix (POROS) anti-HSA column to remove IgG and albumin from plasma respectively. Depleted plasma proteins (50 μL) were diluted 1:4 with binding buffer and subjected to a single chromatographic mixed bed support containing equal volumes of ConA (protein concentration 15 mg/mL and binding capacity of ∼10 mg/mL fetuin and thyroglobulin), Jacalin (protein concentration 15 mg/mL and binding capacity of ∼10 mg/mL fetuin and thyroglobulin) and WGA lectins (protein concentration 15 mg/mL and binding capacity of ∼10 mg/mL fetuin and thyroglobulin) immobilized to POROS matrix beads (total bed volume ∼0.5 mL) (Kullolli et al., 2008). The lectin column was initially washed with 5 column volumes of 50 mM Tris-HCl, 0.5 M NaCl, pH 7.4 (unbound fraction) to elute unbound proteins followed by 5 column volumes of 100 mM acetic acid, pH 4 to elute bound proteins. Both lectin bound and flow-through (FT) fractions were transferred to a C4-reverse phase cartridge to concentrate and desalt the samples. Concentrated proteins were then lyophilized using a vacuum centrifuge and prepared for N- and O-linked oligosaccharide analysis.
SDS Page
Approximately 10 μg of phase partitioned membrane proteins was applied per lane onto NuPAGE 10% Bis-Tris precast gradient gels with MOPS running buffer. Electrophoresis conditions were set to 200 V, 125 mA for 60 min. The gels were fixed in 7% (v/v) acetic acid and 10% (v/v) methanol and stained overnight with Sypro Ruby (Invitrogen, San Diego, CA, USA). Gel images were obtained using Typhoon Trio Variable Mode Imager (GE Healthcare).
Release of N- and O-linked oligosaccharides
N- and O-linked oligosaccharides were sequentially released from lectin-fractionated rat liver membrane and human plasma proteins as described (Wilson et al., 2002) Briefly, Immobilon-P PVDF membranes were prewet with 100% (v/v) ethanol. Rat liver membrane proteins and lyophilized human plasma proteins were resolubilized in 20 μL 8 M urea and dot-blotted (2 × 2.5 μL) on the PVDF membrane and left to dry overnight. PVDF membranes were then washed with methanol followed by water to remove residual salts and detergents. Protein spots were cut from the PVDF membrane and placed in separate wells of a 96-well microtiter plate. The spots were blocked with 100 μL of 1% (w/v) polyvinylpyrrolidone (PVP) 40,000 in 50% (v/v) methanol, agitated for 20 min, and washed thoroughly with water. N-linked oligosaccharides were released from the protein by incubation with 5 μL of PNGase F (0.5 units/μL) for 15 min at 37°C, and subsequently topped up with 10 μL water and incubated overnight at 37°C. The sample wells were surrounded by wells containing water to prevent evaporation, and the plate was sealed tightly with parafilm.
To recover N-linked oligosaccharides, the samples were sonicated in a water bath (in the 96-well plate) for 20 min, and the supernatant containing released N-linked oligosaccharides were collected. Spots were washed twice with 50 μL water, and combined with the supernatant and then reduced to alditols prior to LC separation. Before reduction the released N-linked oligosaccharides were incubated with 100 mM CH3COONH4 pH 5 (final concentration 15 mM) for 1 h at room temperature to ensure complete regeneration of the reducing terminus. N-Linked oligosaccharides were dried under vacuum centrifuge and reduced with 1 M NaBH4/50 mM KOH (20 μL) at 50°C for 3 h. The reduction was quenched with 2 μL glacial acetic acid.
β-Elimination of O-linked oligosaccharides was carried out after the PNGase F release of the N-linked oligosaccharides by rewetting the protein spots with 2 μL methanol, and incubating 16 h with 0.5 M NaBH4/50 mM KOH (20 μL) at 50°C. The reaction was quenched with 2 μL acetic acid and both reduced N- and O-linked oligosaccharides were purified as described below.
Purification of reduced N- and O-linked oligosaccharides
Homemade cation exchange columns comprising 20 μL of AG50W-X8 cation-exchange resin (H+–form) (BioRad, Hercules, CA, USA) were packed on top of μC18 ZipTips (Schulz et al., 2002). Columns were prepared by washing 3 × 50 μL 1 M HCl, followed by 3 × 50 μL methanol and 3 × 50 μL water. N- and O-linked oligosaccharide alditols were eluted with 2 × 60 μL of water and dried. The borate was removed by the addition of 100 μL methanol and drying under vacuum five times. The oligosaccharides were resuspended in 10 μL of 10 mM NH4HCO3, pH 7.8 for Carbon LC–ESI–MS/MS analysis.
Carbon LC-ESI-MS/MS
N- and O-linked oligosaccharide alditols were analyzed by porous graphitized carbon LC-MS/MS on a capillary LC-MS/MS using an Agilent MSD ion-trap XCT Plus mass spectrometer coupled to a Agilent 1100 capillary LC (Agilent Technologies, Placerville, CA, USA). The samples were applied to a Hypercarb porous graphitized carbon column (5 μm Hypercarb, 0.32 × 100 mm, Thermo Hypersil, Runcorn UK). N-linked oligosaccharides were separated using a linear gradient with 2–16% (v/v) ACN/10 mM NH4HCO3 for 45 min, followed by a gradient from 16–45% over 20 min before washing the column with 45% (v/v) ACN/10 mM NH4HCO3 at a flow rate of 5 μL/min for 6 min and reequilibrating in 10 mM NH4HCO3. O-linked oligosaccharides were separated using a linear gradient with 0–25% (v/v) ACN/10 mM NH4HCO3 for 25 min, followed by a 10 min wash with 72% (v/v) ACN/10 mM NH4HCO3 at a flow rate of 5 μL/min. ESI–MS was performed in negative ion mode with two scan events: full scan with mass range m/z 100–2,000 amu and dependent MS/MS scan after collision induced fragmentation. Automated peak recognition and tandem MS of the top three most intense precursor ions at 40% normalized collision energy was performed with an activation time of 30 ms. The composition of glycans was determined using the monoisotopic masses of detected ions and was verified by their associated MS/MS. Monoisotopic masses were searched using the GlycoMod software (available at http://www.expasy.ch/tools/glycomod) with parameters of ± 0.3 Da to predict the possible glycan compositions corresponding to these masses.
Results and Discussion
Rat liver membranes and plasma were chosen to compare the capacity of the same three lectins (ConA, Jacalin, and WGA) to enrich glycoproteins from highly heterogeneous mixtures of hydrophobic and hydrophilic proteins, respectively. In addition, different elution conditions of the bound glycoproteins, via high concentrations of competing monosaccharides (membrane glycoproteins) or by a change in pH (plasma glycoproteins) were used to test for their efficacy in enriching for glycoproteins. The different binding and eluting buffers were necessary because of the particular conditions required to maintain the solubility of the hydrophobic membrane, and hydrophilic plasma, glycoproteins.
Lectin affinity chromatography of standard glycoproteins
We first evaluated the characteristics of the binding of two standard N-linked glycoproteins (mixture of α1-antitrypsin and ovalbumin) to the columns of immobilized ConA, Jacalin, and WGA. α1-Antitrypsin and ovalbumin both contain three possible N-linked glycosylation sites and have not been reported to contain any attached O-glycans. α1-Antitrypsin predominately has attached biantennary complex N-glycans and some triantennary and tetraantennary N-glycan structures (Kolarich et al., 2006). Ovalbumin has mostly high mannose N-glycan structures attached, but also contains hybrid and complex N-glycan structures (Harvey et al., 2000). A mixture of both standard N-glycoproteins (approximately 10 μg of each standard protein) together with an excess of the unglycosylated human serum albumin (HSA) (50 μg) were fractionated on separate lectin chromatography columns containing 0.25 mL of ConA (lectin concentration 10–16 mg/mL and binding capacity 20–45 mg/mL porcine thyroglobulin), 0.5 mL of Jacalin (lectin concentration 5 mg/mL and binding capacity of 1–2 mg/mL human IgA) and 0.5 ml of WGA (lectin concentration 5 mg/mL and binding capacity of 1–2 mg/mL ovomucoid) to test for their retention when present in relatively low abundance. Bound glycoproteins were eluted with 1.5 M methyl-α-glucopyranoside, 1.5 M galactose, and 1.5 M GlcNAc, respectively, to competitively elute the glycoproteins from the lectins. Proteins that flowed through, or were bound and subsequently eluted from the lectin column, were diafiltered, concentrated, and separated by 1D SDS PAGE (Fig. 1).

Lectin affinity fractionation of standard N-linked glycoproteins [mixture of human α1-antitrypsin (10 μg), chicken ovalbumin (10 μg) and human serum albumin (50 μg)]. HSA: human serum albumin (67 kDa), A1PI: α1-antitrypsin (53 kDa) and OVA: chicken ovalbumin (45 kDa). FT: flow through of a mixture of A1PI, OVA, and HSA with ConA (B), Jacalin (D) and WGA columns (F). (B) bound fractions of a mixture of A1PI, OVA and HSA with ConA (C), Jacalin (E), and WGA columns (G).
ConA bound α1-antitrypsin and ovalbumin with mostly HSA in the FT (Fig. 1B and C) and confirms ConA as binding to glycoproteins containing α-mannose in their N-glycans. Both of these N-linked glycoproteins, α1-antitrypsin, and ovalbumin, were not retained well by the Jacalin column although α1-antitrypsin was partially bound (Fig. 1D and E). This suggests that Jacalin, in addition to its primary selectivity for O-glycans [Galβ(1–3)GalNAc], may have some recognition for N-linked glycoproteins through its reported recognition of monosaccharides, including mannose and N-acetylneuraminic acid (NeuAc) (Bourne et al., 2002; Do and Lee, 1998) and its large oligosaccharide binding site (Arockia Jeyaprakash et al., 2005). We have previously observed that Jacalin binds N-linked glycoproteins from a rat liver membrane preparation (Lee et al., 2009). WGA, which recognizes GlcNAc and NeuAcα(2,3), bound α1-antitrypsin and ovalbumin strongly (Fig. 1F and G). The excess unglycosylated human serum albumin (HSA) (50 μg) was shown predominately in the FT fraction of all lectin columns as expected. The presence of a small amount of HSA in the bound fraction could be due to nonspecific interactions to the support media, the lectins, and/or to the two bound glycoproteins.
Lectin affinity chromatography of rat liver membrane and human plasma glycoproteins
Triton X-114 phase partitioned rat liver membrane proteins solubilized in binding buffer containing CHAPS were incubated separately overnight with immobilized ConA, Jacalin, and WGA lectins before mixing and packing into a single tri-lectin column. As seen in Figure 2 some proteins were not bound (FT) but many of the rat liver membrane proteins were retained by the lectin column and were eluted (B) by using competing monosaccharides at a concentration of 1.5 M.
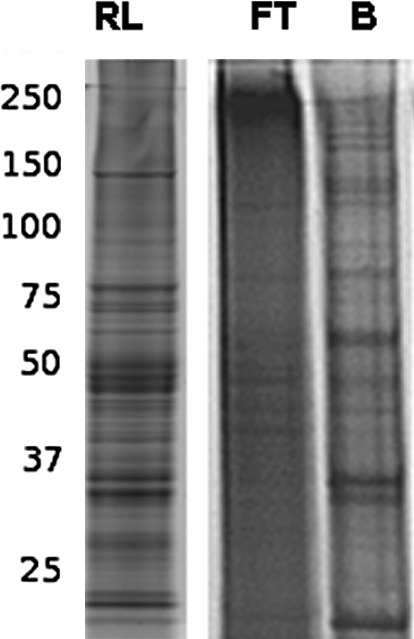
Tri-lectin affinity (ConA, Jacalin, and WGA) bound (B) and flow-through (FT) of Triton X-114 phase partitioned rat liver membrane proteins (RL) eluted from a column containing a mixture of 0.25 mL ConA (10–16 mg/mL), 0.5 mL Jacalin (5 mg/mL), and 0.5 mL WGA (5 mg/mL) with 1.5 M methyl-α-glucopyranoside, 1.5 M galactose, and 1.5 M N-acetyl glucosamine.
Proteomic analysis by mass spectrometry of the tryptic digests of these fractions have previously identified (Lee et al., 2009) that a total of 277 membrane proteins could be enriched by ConA, Jacalin, and WGA lectins, of which 44 (16%) common proteins were isolated by the three lectins when used separately. The identified membrane proteins comprised a large proportion of cell adhesion proteins, receptors, solute carrier transporters, and cytochrome p450 enzymes.
In an alternate approach, albumin and IgG-depleted plasma glycoproteins were fractionated using a single column containing an equal volume of immobilized ConA, Jacalin, and WGA lectins (M-LAC) with each lectin having the same concentration (15 mg/mL). As plasma proteins are water soluble, bound plasma glycoproteins were able to be successfully eluted from the M-LAC column with a low pH buffer. 1D SDS PAGE of the fractionated proteins demonstrated that the bound plasma proteins were largely different to the unbound proteins in the FT from the column (Kullolli et al., 2010).
Although the glycoprotein retention capacity of the amount of lectin used in the column (∼10 mg retention capacity of each as determined by the manufacturer using standard proteins) was far in excess of the total protein sample load (∼100 μg) the possibility of protein saturation of the lectin columns was checked by reinjecting the bound and FT fractions separately back onto the M-LAC column (Kullolli et al., 2010). The same chromatographic behavior of the fractions occurred, that is, the bound fraction was shown to completely rebind to the lectin column and the original FT fraction again did not bind at all to the lectins on the second pass through the column. Thus, the three lectins reproducibly bound to the same glycoproteins and protein saturation of the column was not a contributory factor to the differential protein fractionation by the lectins.
Analysis of N-linked oligosaccharides from lectin-fractionated rat liver membrane and human plasma glycoproteins
ConA and WGA are two of the most commonly used lectins for isolating N-linked glycoproteins. The combination of both these plant-derived lectins has been widely used for improving the analysis of PNGase F released N-linked oligosaccharides from human serum and plasma proteins (Sparbier et al., 2005, 2007) and human colon carcinoma cell membrane proteins (Vercoutter-Edouart et al., 2008). Many plant lectins are themselves N-glycosylated (Kabir, 1995), and extended use of agarose-bound lectins, particularly those conjugated by cyanogen bromide linkages leads to leaching of the immobilized lectins. Thus, plant lectins can sometimes be seen to coelute with the analyzed glycoproteins, and their glycans may contaminate the subsequent analysis. However, the glycans that were derived from any leached lectin proteins can be differentiated from those that were attached to mammalian glycoproteins, because most plant oligosaccharides contain pentose sugars (such as xylose) (Carrington et al., 1985; Kisiel et al., 1996; Kolarich and Altmann, 2000; Wilson et al., 2001), which are not normally found in mammalian N- and O-linked oligosaccharides. In addition, plant oligosaccharides often contain nonmammalian α(1,3) linked core fucose and these will not be released by PNGase F.
To determine how the specificity of the three lectins is reflected in the overall N-glycosylation of the fractionated proteins, N-linked oligosaccharides were released from both the retained and flow through fractions of the lectin-partitioned rat liver membrane and human plasma glycoproteins by PNGase F digestion. The compositions of these N-glycans were determined using graphitized carbon LC–ESI–MS in negative ion mode. The average MS of N-linked oligosaccharides released from lectin-fractionated rat liver membrane proteins is shown in Figure 3, and assigned N-glycan compositions are indicated in Table 1. Surprisingly, the N-glycan global profile of lectin-fractionated rat liver membrane proteins in both the tri-lectin bound and unbound (FT) fractions were remarkably similar. Both show the same relative content of high mannose N-glycan structures [e.g., m/z of (860.3)2− and (941.3)2−)] and sialylated complex N-glycan structures [e.g., m/z of (965.8)2− and (1,111.4)2−].
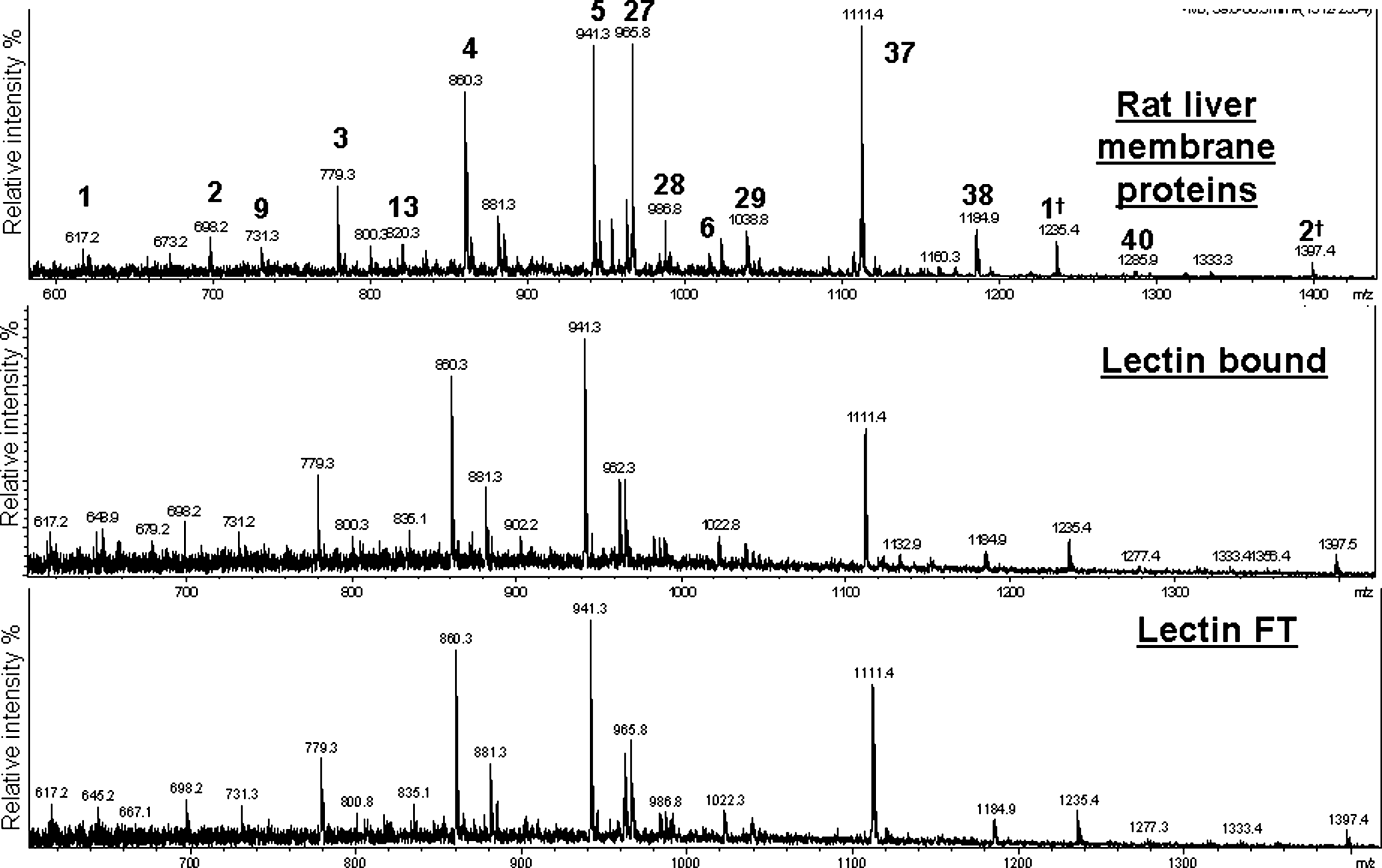
Average MS of released N-linked oligosaccharides (retention time 35–65 min) from rat liver membrane proteins and the tri-lectin (ConA, Jacalin, and WGA) bound and FT fractions. Masses [M-2H]2− corresponding to known rat glycan compositions were observed in all three samples (see Table 1). †Same N-glycan structure but singly charged [M-H]1−.
N-linked core = (Man)3 (GlcNAc)2.
Indicates those N-glycan structures also identified in lectin-fractionated rat liver membrane glycoproteins.
Every effort was made to ensure that the protein load (∼80 μg) was well below the maximum binding capacity of the column [binding capacity 5–10 mg (ConA) and 0.5–1 mg (Jacalin and WGA)] and membrane proteins were solubilized with binding buffer containing CHAPS and filtered prior to incubation with immobilized lectins to minimize the amount of particulates in the sample. It is, of course, possible that membrane proteins may have precipitated during overnight incubation with the lectins and lost their binding specificity.
To try to understand the result of an almost identical N-glycosylation profile of the different proteins that were bound to the lectins compared to those that did not bind, N-glycan profiling of human plasma proteins fractionated by M-LAC (ConA, WGA, and Jacalin) lectin affinity chromatography was also compared. Again, the average MS (over retention time 35–65 min) of the separated N-glycans related from plasma glycoproteins bound to the lectin column (and eluted by a change in pH) and those glycoproteins that did not bind were almost identical (Fig. 4E and K). Both the bound and unbound fractions consisted of high mannose N-glycan structures [e.g., m/z (860.3)2− and (941.3)2−], complex asialo N-glycan structures [e.g., m/z (731.3)2− and (820.3)2−], complex mono-sialo N-glycan structures [e.g., m/z (965.8)2− and (1038.9)2−], complex di-sialo N-glycan structures [e.g., m/z (1,111.4)2− and (1,184.4)2−], and even traces of a complex tri- and tetra-sialo N-glycan structures (Table 1).

Depleted human plasma glycoproteins fractionated by a mixture of immobilized ConA, Jacalin, and WGA lectins. Base peak chromatogram (BPC) of N-linked oligosaccharides released from the bound (
The base peak chromatograms (Fig. 4A and F) of the N-linked glycomic profile were further interrogated in detail to determine if there were any significant glycosylation structural differences on the protein subset that was retained by the three lectins on the column, compared to the glycans on those proteins that were not bound. Graphitized carbon chromatography is able to distinguish and separate isomeric glycans, which may have the same mass and/or composition but differ in specific monosaccharide linkages and thus may exhibit different structural affinities for the lectins.
Using extracted ion chromatograms (EIC) of masses that correspond to specific m/z ions, we selected for the EIC of MS/MS m/z 290.1 (diagnostic ion for NeuAc) (Fig. 4B and G), and m/z 350.2 {diagnostic ion for core fucose [Fucα(1–6)GlcNAc-ol]} (Fig. 4C and H). The EIC of MS/MS m/z 290.1 suggests that the three lectins bound more proteins containing complex sialylated N-glycan isoforms than were in the FT. Conversely, the EIC of MS/MS m/z 350.1 showed that proteins containing core fucosylated N-glycan structures were present in higher abundance in the FT fraction compared to the bound fraction indicating that these three lectins had somewhat reduced affinity to the fucosylated core motif.
To further evaluate the binding of these lectins for specific N-glycan structures, we selected EIC of m/z (1,038.9)2−(Fig. 4D and I), which has the N-glycan composition (Hex)2(HexNAc)2(Fuc)1(NeuAc)1 + (Man)3(GlcNAc)2 and was identified to be a core fucosylated mono-sialylated complex biantennary N-glycan based on the MS/MS fragmentation data. The elution profile of EIC of m/z (1,038.9)2− in the bound and FT fractions showed that two isomeric peaks were eluted at 54 and 61 min, respectively. The peak shown at 54 min corresponds to NeuAc attached in an α(2,6) linkage to the terminus of the complex biantennary N-glycan and the peak shown at 61 min corresponds to NeuAc attached in an α(2,3) linkage to the terminus of the complex biantennary N-glycan structure. The biantennary N-glycan structure consisting of NeuAcα(2,6) was shown to be present in both bound and FT fractions, whereas the N-glycan structure consisting of NeuAcα(2,3) was observed in much greater abundance on the bound proteins than in the FT fraction. The identity of these isomers and their specific NeuAc linkages was confirmed by their having the same MS/MS fragmentation pattern and the same elution position as that of corresponding standard N-glycans (data not shown). Based on the EIC data the combination of ConA, WGA, and Jacalin lectins apparently recognizes specifically N-glycan structures containing terminal NeuAcα(2,3) but not NeuAcα(2,6). In agreement with this observation, the WGA lectin has been reported previously to recognize mostly NeuAcα(2,3) with no recognition for NeuAcα(2,6) and NeuAcα(2,8) (Iskratsch et al., 2009). All three lectins used have been reported not to bind to core fucosylated N-glycan structures (Cummings and Kornfeld, 1982; Iskratsch et al., 2009), which correlates with the predominance of core fucosylated N-glycans in the FT fraction in this study; however, core fucosylated N-glycans with terminal Neuα(2,3) did bind to the M-LAC column (Supplementary Fig. 1). This example demonstrates the complexity of the competing interactions between the heterogeneously glycosylated proteins and the lectin binding affinities.
Analysis of O-linked oligosaccharides from lectin-fractionated rat liver membrane and depleted human plasma glycoproteins
As in the N-linked global glycomic profile, the O-linked oligosaccharide profile on the glycoproteins bound and unbound to the lectins was similar overall. The combined EIC of m/z (675.2)1−, (878.2)1−, (966.3)1−, and (1,331.5)1− (Fig. 5) monitors the masses corresponding to O-glycans previously identified in the rat (Funakoshi and Yamashina, 1982; Hull et al., 1984; Nato et al., 1986) and corresponds to O-glycan compositions consisting of (Hex)1(HexNAc)1(NeuAc)1, (Hex)1 (HexNAc)2(NeuAc)1, (Hex)1(HexNAc)1(NeuAc)2, and (Hex)2 (HexNAc)2(NeuAc)2 as seen in the total liver membrane glycoproteins (Fig. 5A and Table 2). MS/MS fragmentation of the major ions m/z (675.2)1− and (966.3)1− (Fig. 5B and C) found in both the lectin bound and flow through fractions of rat liver membrane glycoproteins identified a mono- and di-sialylated core 1 O-glycan structure (Supplementary Fig. 2 and Table 2).
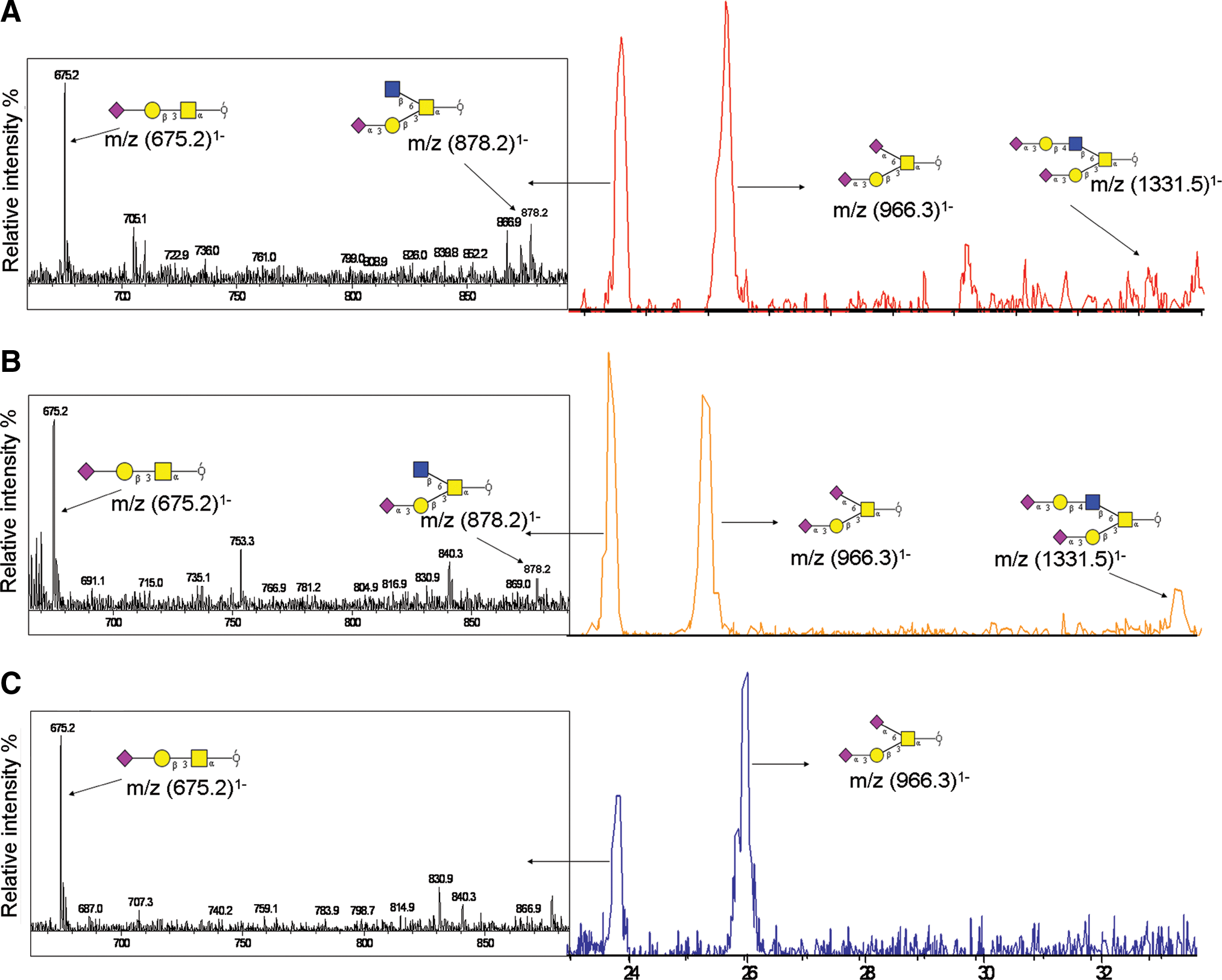
Combined extracted ion chromatogram of m/z (675.2)1−, (878.2)1−, (966.3)1−, and (1,331.5)1− from Triton X-114 phase partitioned rat liver membrane proteins (
 N-acetylgalactosamine
N-acetylgalactosamine  Galactose
Galactose  N-acetylglucosamine
N-acetylglucosamine  N-acetylneuraminic acid
N-acetylneuraminic acid
More detailed analysis of the O-linked oligosaccharides from the lectin-fractionated rat liver membrane glycoproteins was able to show some low abundance differences between the bound and FT fractions. The average MS of masses eluting in the narrow window of retention time 23 to 24 min identified the ions of m/z (878.2)1− and (1331.5)1− in the lectin bound fraction (Fig. 5B) but not in the FT (Fig. 5C). These masses were identified to be mono- and di-sialylated core 2 O-glycan structures by MS/MS fragmentation (Supplementary Fig. 2 and Table 2). The enrichment of these low abundance O-linked oligosaccharides in the lectin bound fraction presumably was due to the preference of the Jacalin lectin for these structures.
O-linked oligosaccharides released from lectin-fractionated human plasma glycoproteins also showed the two major ions of m/z (675.2)1− and (966.3)1−corresponding to the mono- and di-sialylated core 1 O-glycan structures as confirmed by MS/MS (Table 2). These were present in higher abundance in the M-LAC bound fraction compared to the FT (Fig. 6), suggesting that most glycoproteins containing these two O-glycan structures had bound to the M-LAC column.

Base peak chromatogram (BPC) and average MS (retention time 23–27 min) of human plasma proteins fractionated by lectin affinity chromatography shows core 1 O-glycans present in the bound fraction (
These results provide evidence that Jacalin, which is the lectin with preference for binding O-glycans, is suitable for enriching O-linked glycoproteins when used in the multilectin format. Moreover, the inclusion of WGA, that recognizes NeuAcα(2,3), in the tri-lectin mixture, may also assist in the binding and isolation of O-linked glycoproteins containing these sialylated core 1 and core 2 O-glycan structures. The enrichment of O-linked glycoproteins using the combination of the three lectins was consistently observed in both biological systems.
Although using detailed structural analysis there is some preferential binding of specific low abundance oligosaccharide structures to the lectin columns, it is clear, in the case of both the rat liver membrane and the plasma samples, that not all glycoproteins were captured by the tri-lectin column, and that overall, the oligosaccharides on the proteins that were bound to the lectins were mostly the same as those on the proteins that were not retained.
The explanation for this surprising finding is difficult at this stage but we speculate that it may be due to the complexity of interactions that occur in a mixture of the many hundreds of proteins and their glycoforms that comprise these two samples. It has been shown that the tri-lectin M-LAC system has an enhanced affinity for glycoproteins compared to single lectins (Ralin et al., 2008) due to multisite attachment between the glycoproteins and the different lectins. This is supported by the fact that lectins found in vivo exhibit high affinity and specificity for particular glycoproteins presented on cell membranes (Drickamer, 1995). However, protein–protein interactions are considered to be much stronger than the binding of lectins to carbohydrates (Hoebeke et al., 1978; Schreiber, 2002). The dissociation equilibrium constant (Kd) is the propensity for protein complexes to separate reversibly into their components, where lower Kd values indicate stronger binding of a ligand to a particular protein. For example, the affinity of ConA for N-glycans attached to chicken ovalbumin has been reported to have a Kd ∼ 0.34 μM (Ohyama et al., 1985), whereas interactions between proteins such as synapsin I and rat brain synaptic vesicles have been reported to have a lower Kd ∼ 10 nM (Stefani et al., 1997). Therefore, the weaker affinity of lectins for oligosaccharide structures (Kd ∼ μM) compared to the interaction between different proteins in complex mixtures (Kd ∼ nM) may offer one explanation of why similar glycomic profiles are observed on glycoproteins in the lectin bound and unbound fractions.
Conclusions
The data from our previously published proteomic studies (Kullolli et al., 2010; Lee et al., 2009) on the same complex mixtures of rat liver membrane proteins and human plasma proteins as described in this article, showed that the three lectins (ConA, WGA, and Jacalin) reproducibly enriched different subsets of proteins. However, this glycomic study shows, with some exceptions, that similar global oligosaccharide profiles are found on both lectin-bound and -unbound glycoproteins. This suggests that the selectivity of these lectins for specific oligosaccharide structures attached to proteins in complex mixtures appears to be compromised. Such properties as lectin multivalency, the presence of heterogeneous populations of glycans attached to the same protein, the abundance of some structures, the concentration-dependent competing affinities of lectins for the same structures, and/or the competing protein–protein or protein complex interactions may be involved, such that lectin binding cannot simply be explained by their preferred sugar epitopes.
In glycoproteomic studies, lectins are often used and perceived as a method for enriching all intact glycoproteins from complex mixtures. The riddle of lectins presented here, is that the combination of immobilized ConA, Jacalin, and WGA lectins, which are commonly used to enrich N- and O-linked glycoproteins, did not completely capture the entire glycoproteome in rat liver membranes and depleted human plasma, but rather partitioned the proteins into distinct subsets of glycoproteins.
The study emphasizes that the use of lectins in proteomics studies is a valuable tool for the fractionation of different glycoprotein subsets from complex mixtures. The glycomics analysis of these lectin affinity fractionated glycoproteins, however, indicates that both the FT as well as the bound glycoprotein fractions need to be considered for biomarker discovery studies, and that the mechanisms responsible for lectins partitioning complex glycoprotein mixtures might not be the same as those that have been established for purified and well-characterized standard protein mixtures. The almost identical glycomic profiles of unbound and bound glycoproteins cannot be fully explained, but we deduce from our experiments that there appear to be more parameters than simple oligosaccharide recognition that influence the binding (or no binding) of glycoproteins to lectins when complex mixtures are subjected to this kind of enrichment approach.
Footnotes
Acknowledgments
The authors thank their ARC Linkage industry partner GE Healthcare. This research project was supported by access to the Australian Proteome Analysis Facility (APAF) established under the Australian Government's NCRIS program and to the mass spectrometry infrastructure funded by a NSW Cancer Institute Infrastructure grant.
Author Disclosure Statement
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.