
Abstract
Abstract
Growing evidence shows that mutant p53 proteins, which are present in many human tumors, gain oncogenic activities that can actively contribute to tumorigenesis. Mutant p53 proteins have been extensively shown to affect the expression of several genes involved in various aspects of cancer biology. We show here the ChIP-on-chip analysis of mutant p53 binding to a set of 154 promoters, composed of both validated and putative mutant p53 target genes. By using the chromatin obtained from mutant p53R175H-immunoprecipitation in proliferating SKBr3 breast cancer cells, we found that mutant p53 binds to 40 of the 154 promoters analyzed. siRNA-mediated mutant p53 knock-down modulates the transcript abundance of some of these target genes. Two-thirds of the mutant p53-bound promoters were also engaged by either p300 or PCAF acetyl-transferases, strongly indicating the presence of transcriptionally active complexes. We also found that NF-kB binding sites are overrepresented among the mutant p53-bound promoters; a ChIP-on-chip analysis confirmed that NF-kB p65 binds to 27 of the mutant p53-bound promoters, indicating that mutant p53 could influence the transcriptional output of these NF-kB target genes.
Introduction
The molecular mechanisms underlying gain of function of mutant p53 proteins still remain to be fully elucidated. Two-thirds of missense mutations in the DNA-binding domain, including all hotspot mutations, abrogate the ability of p53 to bind and transactivate canonical wild-type p53 binding sites; despite that, mutant p53 proteins have been shown to modulate gene expression and were recently found recruited to the regulatory regions of genes with important roles in cancer biology. These findings raise the possibility that mutant p53 functions as an oncogenic transcription factor capable to aberrantly modulate gene expression (reviewed in Strano et al., 2007). A unifying mechanism for the selectivity of mutant p53 toward certain genes is still missing, owing to the lack of a consensus DNA sequence among genes regulated by mutant p53 and the variability in the identity of the genes affected by different p53 mutants. We and others have previously reported that mutant p53 reaches target gene promoters through the interaction with sequence-specific transcription factors, such as NF-Y, E2F1, NF-kB, and Vitamin D receptor (Di Agostino et al., 2006; Fontemaggi et al., 2009, 2010; Stambolsky et al., 2010; Weisz et al., 2007). Mutant p53 was shown to bind to NF-YA, which augments its recruitment to the promoters of cell-cycle regulatory genes following treatment with chemotherapy agents; this results in the aberrant transcriptional activation of genes involved in the control of G2/M transition, such as Cdk1 and cyclin B2, upon exposure to DNA damaging agents (Di Agostino et al., 2006). Unlike the mutant p53/NF-YA interaction, which is observable only after DNA damage, we have recently shown that E2F1 cooperates with endogenous mutant p53 in the transactivation of the ID4 (inhibitor of DNA binding 4) promoter in proliferating breast cancer cells under basal conditions; mutant p53 and ID4 protein coexpression lead to an increase in the angiogenic potential of these cells (Fontemaggi et al., 2009). Besides E2F1, we found that NF-kB also carries mutant p53 to the ID4 promoter. A functional crosstalk between NF-kB and mutant p53 had been also previously reported by Weisz et al. (2007), who found that mutant p53 can augment the induction of NF-kB transcriptional activity in response to tumor necrosis factor (TNF)-α. Very recently, mutant p53 was also found to differentially modulate subsets of VDR target genes and to convert vitamin D into an antiapoptotic agent (Stambolsky et al., 2010).
In the present study we examined, by ChIP–chip approach, the ability of one of the most frequent cancer-associated p53 mutants, p53R175H, to interact with the promoters of an array of already validated or putative mutant p53 target genes. We identified 40 promoters that are involved in interactions with p53R175H in breast cancer-derived SKBr3 cells. The analysis of p300 and PCAF acetyltransferases recruitment to the mutp53-bound promoters confirmed a subset of promoters carrying transcriptionally active protein complexes. Finally, a bioinformatics analysis indicated that the mutp53-bound promoters are enriched for NF-kB binding sites, relative to all annotated promoters; ChIP-on-chip data confirmed the presence of p65 NF-kB on 27 of those. Together, these data highlight that mutant p53 is recruited to a subset of its target promoters even in the absence of stimulating agents and that the transcription factor NF-kB is one of the major mediators of mutp53 binding to its target promoters.
Materials and Methods
Cell cultures
SKBr3 breast cancer cells were cultured in D-MEM medium with 10% (v/v) FCS plus 2 mM L-glutamine and antibiotics.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed basically as described previously (Fontemaggi et al., 2001). Chromatin from exponentially growing SKBr3 cells was precleared with protein G agarose beads (Pierce, Rockford, IL, USA) for 1–2 h at 4°C and subsequently incubated overnight with the following antibodies: p53 (sheep polyclonal Ab-7; Calbiochem, LaJolla, CA, USA), p53 (FL-393; Santa Cruz, Santa Cruz, CA, USA), p300 (Santa Cruz, C20 and N15), PCAF (Santa Cruz, E-8) and p65 NF-kB (Santa Cruz, sc-372). The next day protein G beads (Pierce) preblocked overnight with 1 μg/μL bovine serum albumin (BSA) and 1 μg/μL sheared herring sperm DNA were employed to finally immunoprecipitate the protein–DNA complexes. After reversal of formaldehyde crosslinking, RNase A, and Proteinase K treatments, IP enriched DNAs and input DNAs were used to generate amplicons for hybridization experiments. Independent ChIP experiments with subsequent semiquantitative and quantitative (SYBR Green assay) polymerase chain reaction (PCR) were used to validate the results of the ChIP-on-chip analysis. Primer sequences used for the PCR reactions of BCL2L1, FAS, cFLAR, BIRC3, BIRC2, ARTS1_2, ARTS1_3 are available upon request.
ChIP-on-chip
To obtain sufficient amount of IP DNA (1 μg) for the microarray hybridization, we performed ligation-mediated PCR (LM-PCR) as described earlier (Oberley et al., 2004). In brief, two unidirectional primers were annealed and ligated to the chromatin of the IPs and to the input DNA as an internal reference, previously blunted by T4 DNA polymerase. The resultant DNA population becomes amplified by PCR and, after a purification step, quantified. Subsequently the amplicons were labeled indirectly by incorporating the nucleotide analogue aminoallyl dUTP (Fermentas, Hanover, MD, USA) by random priming into the DNA (BioPrime; Invitrogen, Carlsbad, CA, USA), followed by the conjugation of the fluorophor (Cy3 or Cy5, GE Healthcare-Amersham, Piscataway, NJ, USA) to the incorporated nucleotide analogue. The experimental amplicon became labelled with Cy5, the input amplicon with Cy3.
The slides to be hybridized were generated by spotting oligonucleotides (0.5 μg/μL) onto gamma aminopropyl silane coated slides (Corning slides and spotting buffer Pronto!TM Universal; MicroGRID II plus compact spotter; Genomic Solutions). Each oligonucleotide was represented 18 times, distributed on the array. The slides were prehybridized for 2 h at 55°C with hybridization buffer (Genesphere, buffer 6) supplemented with 10 ng/μL sheared salmon sperm DNA. After washing, the arrays became cohybridized with ChIP Cy5- and input Cy3-labeled amplicons for 16 h at 55°C. The slides were washed 5 min with 1× SSC 0.1% SDS, preheated to 55°C, followed by several washing steps: 5 min 1× SSC 0.1% SDS 42°C, 5 min 0.5× SSC 37°C, 5 min 0.2× SSC 37°C, 5 min 0.1× SSC 55°C, and 5 min 0.1× SSC at room temperature (RT). Finally, the slides were spun dry, scanned in the 4200A Axon scanner, and analyzed with the GenePix Pro® 6.1 software. For data normalization and analysis for each ChIP–chip experiment three different samples were prepared and used in two different hybridization comparisons: specific IP versus input and nonspecific IP (no antibody) versus input. The second quartile of lowest intensities served for normalizing the two slides (specific ChIP slide vs. no antibody ChIP slide). The fold enrichment was determined by comparing the ratio of the specific ChIP Cy5 versus input Cy3 to the ratio of no antibody control ChIP Cy5 versus input Cy3.
RNA interference
Si p53 oligonucleotides contain the sequence published by Brummelkamp et al. (2002). siScramble contains the following scrambled sequence of p53 siRNAs: CTA TAA CGG CGC TCG ATA T.
RNA extraction and RT-PCR
Total RNA was extracted using the Trizol Reagent (Gibco BRL, Gaithersburg, MD, USA) following the manufacturer's instructions. The first-strand cDNA was synthetized according to manufacturer's instructions (M-MLV RT kit; Invitrogen). mRNA expression were measured by PCR. Primer sequences for HIRA, NFS1, LIG1, PCDHA7, CD95, RBBP6, TGFB2, GAPDH are available upon request.
Results
Mutant p53R175H binds to 40 selected target promoters
It is well established that mutant p53 cannot bind to the consensus sequences of wtp53 on the regulatory regions of p53 target genes. Nevertheless, several experimental evidences indicate that one of the mechanisms underlying mutant p53 GOF is the modulation of gene expression. Indeed, mutant p53 proteins can be recruited to the promoters of their target genes.
We wanted to explore to what extent mutant p53 modulates gene expression through the direct recruitment of target gene promoters. To this end, the Chromatin Immunoprecipitation (ChIP)–chip approach was undertaken to evaluate in vivo binding of mutant p53 protein to the promoters of its target genes. We designed and produced a low-density slide (mutp53-chip) carrying oligonucleotides that correspond to the promoter regions of 154 putative and proven target genes of mutant p53. The list of promoters (Supplementary Table 1) was generated by choosing those genes that: (1) were already shown in the literature to have some dependency upon mutant p53 expression (reviewed in Oren and Rotter, 2010); (2) were found to be bound by mutp53 in a previous ChIP–chip analysis (Stambolsky et al., 2010); (3) were found modulated by mutant p53 in a previously performed expression microarray analysis of mutant p53 inducible cells (Fontemaggi et al., 2009). Each promoter is represented on the slide by three 50-mer oligonucleotides, derived from around positions −800, +1, and +800 relative to the transcription start site.
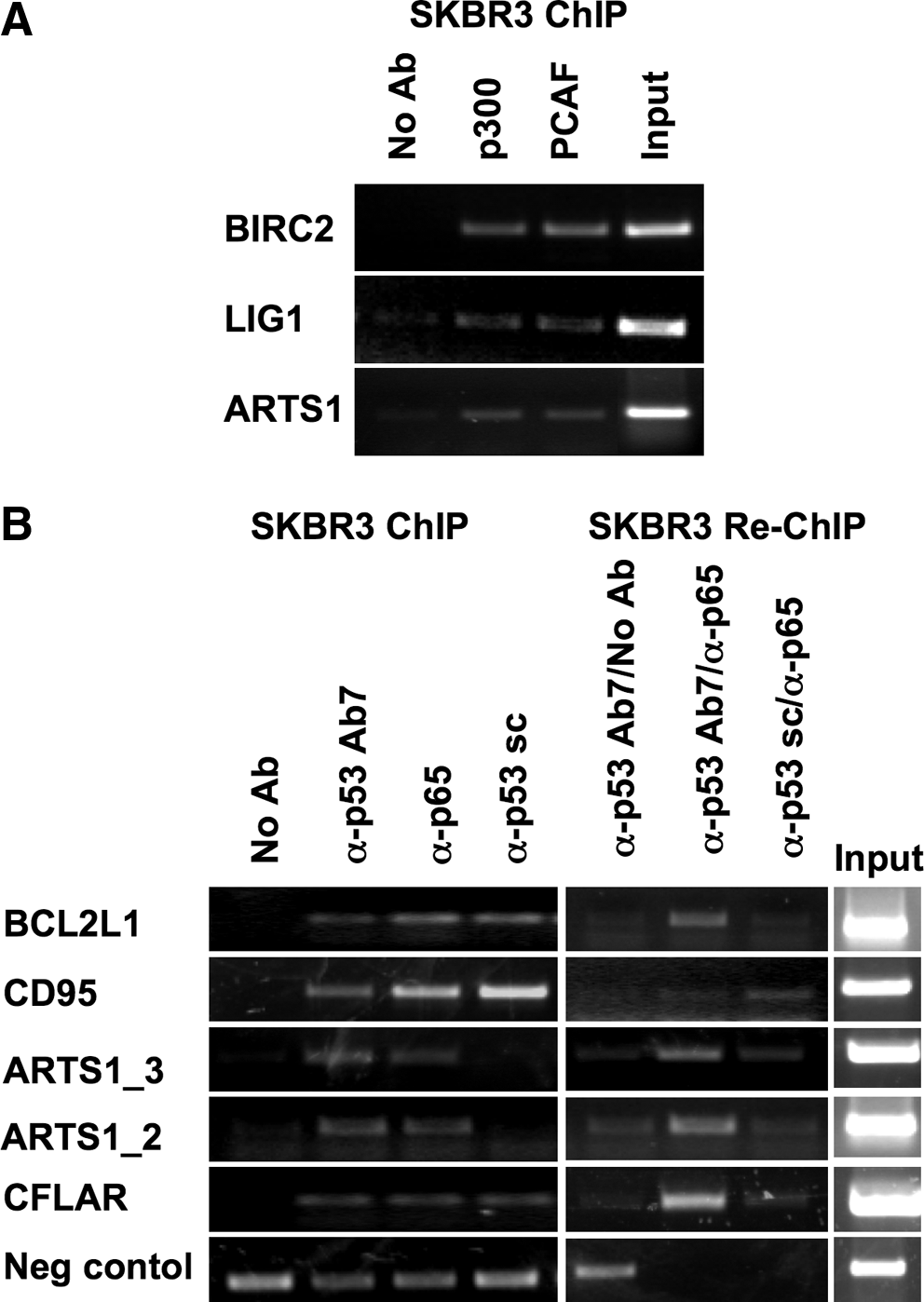
We performed ChIP for p53 in proliferating SKBr3 breast cancer cells, endogenously expressing mutant p53R175H. The obtained DNA was amplified, labeled, and hybridized to the mutp53-chip, and enrichments versus the negative control sample (no antibody) were evaluated. We found that mutant p53 binds to the promoters of 40 out of 154 genes (Table 1); the enrichments were present for one probe in 34 promoters and for two probes in 6 promoters (see Table 1). The binding of mutant p53R175H to five randomly selected promoters from among this group was confirmed by additional ChIP experiments and is shown in Figure 1A.

Mutant p53 associates with 40 of the promoters represented on the ChIP–chip slide. (
1,2 value was chosen as cutoff for enrichments.
ChIP–chip analysis allowed us to identify several novel mutant p53 direct target genes, each of which could potentially contribute to the various facets that characterize mutant p53 GOF. To explore the biological role of mutant p53 targets we performed a functional classification. As shown in Figure 1B, more than half of the genes found to be bound by mutant p53 are involved in the control of apoptosis or inflammation.
To evaluate the impact of mutant p53 binding on target gene expression, we knocked down mutant p53 expression in SKBr3 cells and performed RT-PCR analysis for some transcripts. As shown in Figure 1C, among seven transcripts analyzed, four were downregulated (HIRA, NFS1, LIG1, PCDHA7), one (CD95/FAS) was upregulated, and two were not detectably modulated (RBP6, TGFB2) following p53 silencing. These results indicate that mutant p53 can bind to promoters and modulate gene expression positively or negatively. The fact that we also found genes bound by mutp53 but unaffected by mutp53 depletion suggests that mutant p53 transcriptional activity might be exerted on these promoters only after specific stimuli (e.g., chemotherapy treatment).
Several of the genes included in the mutp53-chip design are known to be regulated by both wild-type and mutant p53 proteins, mostly in opposite ways; one example is FAS (CD95), which is upregulated by wtp53 (Muller et al., 1998; Owen-Schaub et al., 1995) and downregulated by mutant p53 (Zalcenstein et al., 2003). These opposite activities seem to rely on different regulatory regions within the target gene, with wtp53 often associating with consensus p53 binding sites located in the first intron of the target gene and mutant p53, on the contrary, binding the promoter region through the mediation of other transcription factors. Accordingly, the distribution analysis of wtp53 binding sites on the 40 promoters bound by mutant p53, performed using RegionMiner (Genomatix, Munich, Germany), did not yield any significant enrichment for wt p53 consensus sites when considering regions of ± 200 bp or ± 300 bp around mutp53-positive probes (Supplementary Table 2). Also, the hybridization of the mutp53 slide with the DNA obtained from wt p53-immunoprecipitated chromatin, derived from MCF-7 breast cancer cells, revealed that 12 out of 154 promoters were recruited by wtp53 and, among those, only 4 were also bound by mutant p53 in SKBr3 cells (Table 1 and Supplementary >Table 3). These results confirm the specificity diversity of the binding of mutant p53 to selected target promoters.
Mutant p53 takes part in transcriptionally active protein complexes
To investigate whether the binding of mutant p53 to a given target promoter corresponds to an active role in transcriptional control, we analyzed the recruitment of p300 and PCAF acetyltransferases to the promoters represented on the slide. p300 was previously shown to contribute to mutant p53 transcriptional activity mediated by either NF-Y or E2F1 (Di Agostino et al., 2006; Fontemaggi et al., 2009). Notably, 24 out of 40 mutant p53-bound promoters were found to also be engaged by either p300 or PCAF (Table 2). The binding of p300 and pCAF to the BIRC2, LIG1 and ARTS-1 promoters was verified on an independent ChIP experiment (Fig. 2A).
(
Mutant p53 and p65 (NF-kB) bind concomitantly to selected target promoters
Despite extensive experimental efforts, evidence that GOF mutant p53 proteins bind to target gene promoters through a specific DNA binding consensus has so far not been obtained. Previous work from our group has shown that mutant p53 can be recruited to target promoters through cooperation with sequence-specific transcription factors, like NF-Y, NF-kB, and E2F1 (Di Agostino et al., 2006; Fontemaggi et al., 2009).
As expected, a computational search for common sequences on mutant p53-bound promoters, in attempt to identify a common mutant p53 binding consensus, did not yield any positive result (data not shown). We next performed a distribution analysis of transcription factor binding sites, considering regions of ± 200 or ± 300 bp around the positive probes of mutant p53-bound promoters using RegionMiner (Genomatix).
As shown in Table 3, we found a significant enrichment for NF-kB consensus binding sites on mutant p53-bound regions, both when the analysis was performed versus the genome (overrepresentation: 2.39) and, more importantly, versus annotated promoters (overrepresentation: 1.85, Z-score: 2.65). A functional crosstalk between NF-kB and mutant p53 has been demonstrated by Weisz et al. (2007), who reported that mutant p53 can augment the induction of NF-kB transcriptional activity in response to TNF-α. We asked whether mutant p53 could reach some of its target promoters through the cooperation with NF-kB even in the absence of TNF-α. To that end, we immunoprecipitated SKBr3 chromatin with an antibody directed to the p65 (RelA) subunit of the NF-kB complex and the obtained DNA was hybridized to the mutant p53 chip. We found that p65 binds to 86 promoters, among which 27 are shared with mutant p53 (Supplementary Table 4). The binding of p65, as well as that of mutant p53, to some of the shared target promoters were confirmed by additional ChIP experiments, as shown in Figure 2B (left panel). To investigate whether there is a concomitant presence of mutant p53 and p65 on the same genomic region, indicating the formation of transcriptionally competent complexes containing both proteins within the same complex, we performed Re-ChIP analyses. As shown in Figure 2B (right panel), we found that mutant p53 and p65 are indeed simultaneously present on the promoters of ARTS1, BCL2L1, FAS, and CFLAR.
Discussion
In the present study we used ChIP-on-chip to analyze the ability of mutant p53 to directly control the expression of an array of validated and putative target genes. For this purpose we generated a low-density array, the mutp53-slide, containing probes corresponding to the regulatory regions of 154 promoters. By using the mutp53-slide we identified the presence of mutant p53R175H, endogenously expressed in SKBr3 breast cancer cells, on 40 of 154 analyzed promoters. When we analyzed some of these transcripts by RT-qPCR we found both genes whose expression was up- or down-modulated in the absence of mutp53 and genes that were not affected by mutp53 depletion. This indicates that mutp53 recruitment to target promoters does not necessarily correlate with an active transcriptional state. We hypothesize that mutp53 is bound to some target promoters but becomes transcriptionally active only after specific stimuli. This is a completely unexplored field that surely deserves further investigation. Increasing evidence indicates that mutant p53 reaches chromatin regions by association with sequence-specific transcription factors, leading to an aberrant transcriptional modulation of their target genes. This has been demonstrated for NF-Y, E2F1, and VDR (Di Aostino et al., 2006; Fontemaggi et al., 2009; Stambolsky et al., 2010). Mutant p53 was also found to drive the activity of the NF-kB complex in response to TNF-α, attenuating TNF-α-induced apoptosis (Weisz et al., 2007). The mutp53 slide represents a useful tool for the characterization of the protein complexes that enable mutant p53 to reach its target promoters. By analyzing the presence of transcription factor binding sites on the 40 promoters bound by mutp53, we found a significant overrepresentation of the NF-kB consensus sequences. ChIP–chip analysis of p65 recruitment to these promoters confirmed that indeed about 60% of the identified mutp53 targets are also bound by p65. A Re-ChIP analysis on a restricted group of these targets indicated that mutp53 and p65 are concomitantly recruited to these promoters. As mentioned above, a functional cooperation was previously demonstrated to occur between mutp53 and NF-kB in response to TNF-α (Weisz et al., 2007). Their concomitant recruitment on a target promoter was also successively demonstrated for the ID4 promoter by ChIP analysis (Fontemaggi et al., 2009). Collectively, our findings have several important implications: (1) the range of mutant p53 target genes can be very broad, in accordance with the number of the transcription factors to which mutant p53 can physically associate; (2) NF-kB might represent a major mediator of the DNA-binding activity of mutp53; (3) transcriptional activity of mutant p53 might occur even in the absence of any external stimuli, thereby contributing to the maintenance of the transformed phenotype of mutant p53-expressing cancer cells.
Conclusions
The characterization of the protein complexes that enable the recruitment of mutant p53 proteins to the regulatory regions of target genes is of crucial importance for deciphering the mutp53 GOF mechanisms. The low-density microarray slide used in the present study is a powerful method for the analysis of such complexes. Our results indeed indicate an overrepresentation of NF-kB consensus sequences on mutant p53 target promoters in SKBr3 cells, which harbor mutant p53R175H. The future investigation of the protein complexes encompassing mutant p53, in cell lines derived from various tissues and carrying other types of mutations, under basal conditions and in response to specific stimuli, will shed further light on the involvement of other transcription factors as mediators of mutant p53 transcriptional activity. This gained knowledge might also be useful toward deciphering the epigenetic events underlying transcriptional GOF activity of mutant p53 proteins, thereby providing molecular evidence for novel epigenetic cancer approaches.
Footnotes
Acknowledgments
This work was supported by the European Community (EC) FP6 “Active p53” consortia. This publication reflects the authors' views and not necessarily those of the EC. The EC is not liable for any use that may be made of the information contained herein. The support given by AIRC-ROC to the oncogenomic platform, AIRC to G.B. and S.S., Lega Italiana Tumori to S.S., Fondazione Veronesi, Ministero della Sanità, and Alleanza contro il cancro is greatly appreciated.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.