
Abstract
Abstract
Currently available technologies allow in-depth analysis of multiple facets of the proteome that have clinical relevance and that complement current genomics-based approaches. Although some progress has been made in our knowledge of the human proteome in health and in disease, there is an urgent need to chart a coherent road map with clearly defined milestones to guide proteomics efforts. Areas of emphasis include: (1) building resources, (2) filling gaps in our understanding of biological variation, and (3) systematically characterizing proteome alterations that occur in well-defined disease states, all of which require an organized and collaborative effort.
Introduction
Range of proteomic interrogations that complement genomic-based studies.
Proteomics technologies currently allow identification of a near exhaustive list of proteins for which an mRNA is transcribed in a given cell population. For example, in a study of pancreatic islets of Langerhans 6,873 proteins were identified (Waanders et al., 2009). Quantitative comparison of the proteome of single islets, containing 2,000–4,000 cells, treated with high or low glucose levels, allowed analysis of a repertoire of proteins involved in diabetes, including novel components. At the subcellular level, the secretome, the cell surface proteome, and the phosphoproteome can be analyzed with exquisite detail using proteomics technologies to identify potential novel diagnostic and therapeutic targets (Chenau et al., 2009; Daub et al., 2008; Faca et al., 2009; Greco et al., 2010; Gundry et al., 2008; Kohnke et al., 2009; Kosako et al., 2009; May, 2009; Piersma et al., 2010; Rinschen et al., 2010; Xu et al., 2010; Xue et al., 2010). Likewise, proteome profiling of tissues has allowed searches for disease alterations in great depths (Beretta, 2009; de la Cuesta et al., 2009; Voshol et al., 2009; Parikh et al., 2010; Sanchez-Carbayo, 2010).
Depth of Analysis of the Serum and Plasma Proteomes
Interest in profiling serum, and plasma from which serum is derived, using proteomics has been of long standing. The serum and plasma proteomes have been the subject of numerous studies in part due their importance as sources of disease biomarkers for most common diseases (Blennow et al., 2010; Chen et al., 2010; Hanash et al., 2008; Kijanka et al., 2009; Paczesny et al., 2009; Somasundaram et al., 2009; Whiteley et al., 2009). Approaches that have been successful in improving our understanding of the proteome and elucidating alterations associated with disease have aimed at decomplexing the proteome into manageable components that are investigated separately, through “divide-and-conquer” strategies. Such strategies are particularly pertinent to plasma given the vast dynamic range of protein abundance in this biological fluid, spanning many orders of magnitude and given the multiplicity of features associated with plasma proteins (Apweiler et al., 2009).
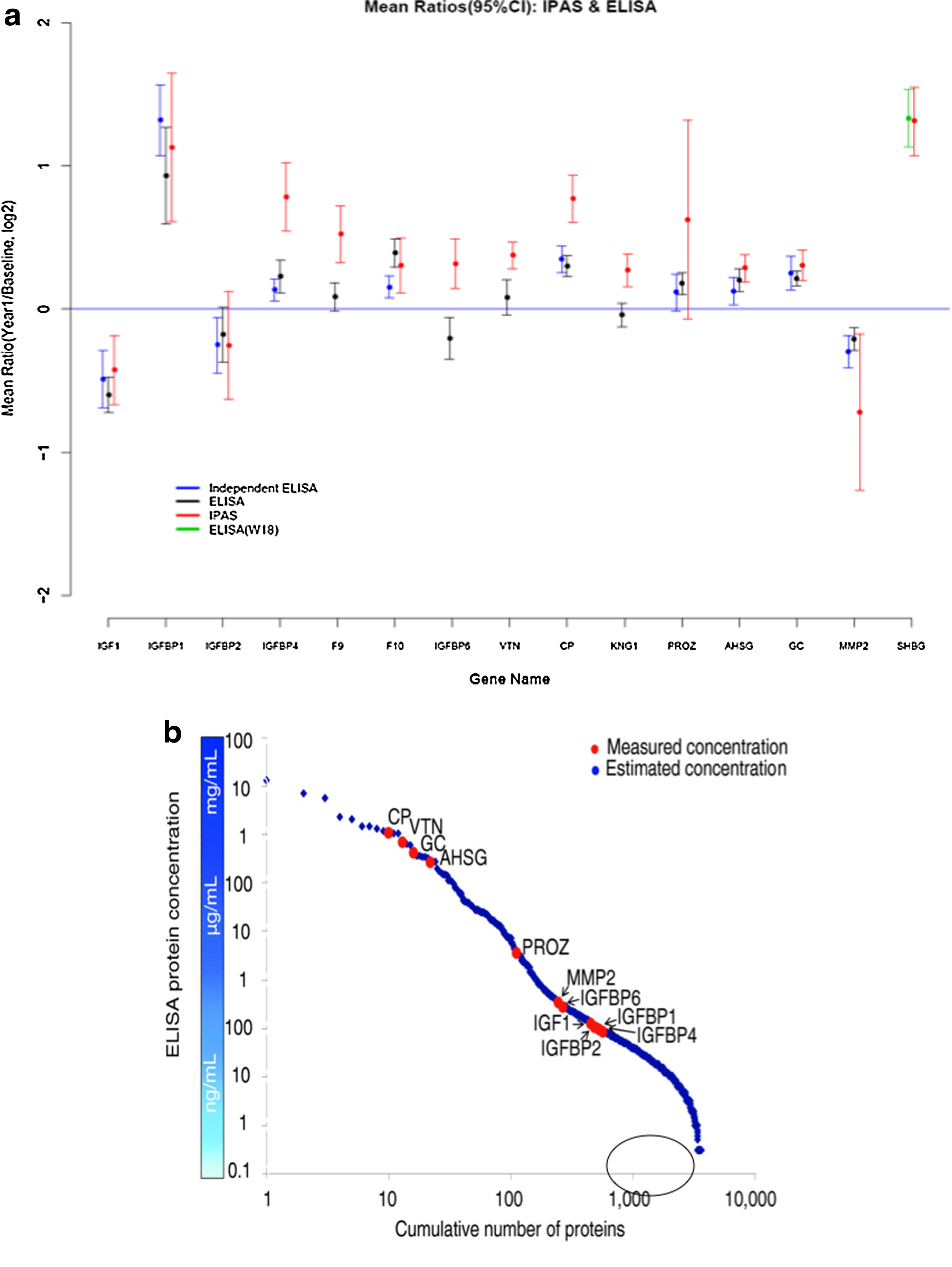
We have engaged in a project to determine the depth that may be achieved in analysis of the serum proteome through a protein fractionation strategy, followed by tryptic digestion and LCMSMS analysis of individual fractions, and subsequent integration of resulting MS data (Faca et al., 2007). From a biological point of view, the objective of the project was to determine the impact of a particular therapy, namely, postmenopausal estrogen and progestin treatment on the serum proteome and the potential to identify risk factors and markers for common diseases (Katayama et al., 2009). A comparison was made of the serum proteome of 50 healthy women at baseline and at 1 year following randomization to postmenopausal estrogen therapy. Comparisons between serum at baseline and serum at 1-year postestrogen therapy were made based on differential isotopic labeling with heavy and light acrylamide prior to mixing and fractionation. In this initial study, 1,760,094 spectra yielded 1,612 proteins identified with a <5% error rate. A remarkable 19% of quantified proteins met a 0.05 significance level criterion for change after 1 year of estrogen therapy indicative of the extent to which serum and plasma may reflect physiologic states and may be impacted by various exposures. Equally remarkable is the concordance between mass spectrometry based quantitative data and ELISA data for proteins subjected to validation using serum aliquots from the same group of subjects as well as serum aliquots from an independent set of women that received the same therapy but were not subjected to mass spectrometry (Fig. 2a). The correlation between mass spectrometry based proteomic log-ratios of concentration differences between baseline and year 1 and ELISA log-ratios was strong (correlation = 0.83). Of interest, the measured abundance range of the quantified proteins some of which were subjected to ELISA, spanned more than six orders of magnitude, indicative of the depth of proteomic analysis that was achieved (Fig. 2b).
(
Postmenopausal hormone therapy with estrogen (E) or with estrogen plus progestin (E + P) has been shown to have multiple effects of public health importance (Anderson et al., 2004; Rossouw et al., 2002). In some studies, the observed effects were similar for the two preparations for some outcomes, including stroke (Hendrix et al., 2006; Wassertheil-Smoller et al., 2003) and hip fracture (Cauley et al., 2003; Jackson et al., 2006), whereas E + P effects were unfavorable compared to those for E-alone for other outcomes, including coronary heart disease (CHD) (Hsia et al., 2006; Manson et al., 2003), breast cancer (Chlebowski et al., 2003; Stefanick et al., 2006), and venous thromboembolism (VT) (Curb et al., 2006; Cushman et al., 2004). The spectrum of biological changes induced by hormone therapy in relation to the outcome effects has remained unclear. So we compared the serum proteome at 1 year following randomization to baseline for 50 women assigned to E + P and for 50 women assigned to E-alone (Pitteri et al., 2009). Quantitative changes were found to be highly correlated between the two hormone therapy preparations and included multiple proteins relevant to coagulation, inflammation, immune response, metabolism, cell adhesion, growth factors, and osteogenesis. There was also evidence of differential changes between the two hormone preparations, particularly for some growth factors, and inflammatory pathways may be related to the differential risk for breast cancer between the two preparations.
The Need for Organized Collaborative Approaches to Synergize Effort in Clinical Proteomics
These case studies of serum proteomics are but examples that illustrate the depth of coverage and potential insights into disease that may be uniquely gained from proteomic analysis even with current technologies. Ever since the emergence of proteomics as a field more than a decade ago, there has been a need for an organized effort in proteomics (Hanash, 2004). Given the success of the genome project, it was felt that likewise, there should be a proteome project. However, there are substantial differences between an undertaking to sequence the human genome and an undertaking that tackles the human proteome in all its aspects. It is therefore impractical to conceptualize an all-encompassing human proteome project. An alternative would be to conceptualize several targeted, well-focused proteome projects and initiatives with clearly defined objectives and milestones. Proteome projects may be clustered into three groups: (1) building resources; (2) elucidation biological variation at the proteome level, and (3) systematically characterizing proteome alterations that occur in well-defined disease states.
Building Resources for Proteomics
The Human Proteome Organization (www.HUPO.org) came into existence to help identify needed resources and determine achievable objectives to further our understanding of the human proteome (Service, 2003). Early on, HUPO organized numerous meetings in North America, Asia, and Europe, with participation by government and industry representatives and academicians to help define needed resources and prioritize objectives. As an example, a substantial need was identified for informatics resources for practically every aspect of proteomics. Approaches to the analysis of protein data have been highly informal and nonstandardized. An important informatics-related effort was initiated that was aimed at developing and adopting standardized approaches to facilitate analysis of proteomics data generated by different laboratories. Thus, the Proteomics Standards Initiative (PSI) was launched with the aim of defining community standards for data representation in proteomics to facilitate data comparison, exchange, and validation (Hermjakob et al., 2006). The PSI has been a success and has endeavored to develop standards and tools that make proteomics data accessible. Proteomics experiments can result in vast quantities of data that require efficient tools for their analysis. The latest release from PSI is a platform independent jmzML, a Java Application Programming Interface for mzML files that combines a small memory footprint with a fully functional object model without sacrificing the speed of data access which typifies the need for standards (Cote et al., 2010). Such standards are increasingly being adopted (Martinez-Bartolome et al., 2010).
Aside from protein standard-related initiatives, there have been numerous proposals to develop various types of resources for proteomics. A notable initiative is Protein Atlas (Berglund et al., 2008). This project aims to experimentally annotate the human protein complement of the genome in a gene centric manner. The approach relies on the use of antibodies to design protein-specific probes for a representative protein from every protein-coding gene. Such antibodies are used for systematic analysis of cellular distribution and subcellular localization of proteins in normal and disease tissues. Profiles have been generated for a large fraction of the proteins encoded in the human genome. Validation performed for antibodies include a protein array assay, Western blot analysis, immunohistochemistry, and immunofluorescence-based confocal microscopy. The protein atlas is representative of resources needed for proteomics to impact many areas of biomedical research. An application of Protein Atlas is the development of a Web-based tool for in silico biomarker discovery related to cancer (Bjorling et al., 2008). Search queries are based on the human tissue profiles in normal and cancer cells in the Human Protein Atlas portal and rely on annotations performed by pathologists of images representing immunohistochemically stained tissue sections. Search tools allow for exploration of the protein atlas to discover potential tissue-specific, cell type-specific, and tumor type-specific markers.
Some effort has been made to address needs for resources through government initiatives. However, such effort falls far too short to meet the pressing needs for resources in proteomics. Recently, HUPO has proposed a gene-centric approach to produce a human proteome map that contains information about the proteins expressed from each known gene locus and to make this information publicly available (2010). A three-part approach was proposed, that can be implemented with current technology, such that for each predicted protein-coding gene, at least one of its major representative proteins would be characterized as to the major tissues where it is expressed, its abundance, and its interacting protein partners. Other projects have been proposed that vary in scale and that address the need for particular resources such synthetic peptides and affinity capture agents to assist in proteomic studies. What appears to be needed at the present time in addition to well-conceived execution plans and distribution of efforts and milestones, is a substantial buy-in for these initiatives and projects from individuals from outside the proteomics community that see a clear benefit from such projects for their own areas of interest.
Elucidation of Biological Variation at the Proteome Level
A prerequisite to elucidating alterations associated with disease is to understand the extent of biological variation in the proteome that occurs in individuals under physiologic states, and among individuals and populations. Understanding biological variation should be the subject of well-designed collaborative projects resulting in publicly available data resources. The relevance of such projects is illustrated in a limited study aimed at identifying quantitative trait loci underlying proteome variation in human lymphoblastoid cells (Garge et al., 2010). Expression levels of 544 proteins in a population of 24 individual human lymphoblastoid cell lines (LCLs) that have been extensively genotyped as part of the International HapMap Project were determined. A subset of 15 proteins were observed for which genetic elements were responsible for >50% of the expression variation. The genetic variation associated with protein expression levels were located in cis with the gene coding for the protein's transcript for most of these proteins. Four of the genetic elements identified were coding nonsynonymous single nucleotide polymorphisms (NS-SNPs) that resulted in changes in the migration pattern of the corresponding proteins.
The first proteome project to be conceived through HUPO activities is the Plasma Proteome Project (Hanash, 2004), which included an objective to study biological variation and which resulted in completion of a pilot phase. There was consensus among leaders in industry, government, and academia that the plasma is perhaps the most crucial compartment to inform about the status of health and disease of an individual through profiling of its protein constituents. Several workshops were held to help define the project. The scientific objectives of a plasma proteome project (Table 1) largely as outlined nearly a decade ago still remain highly meritorious.
The pilot phase had several objectives: (1) compare a broad range of technology platforms for the characterization of proteins in human plasma and serum, including in particular low abundance proteins and assess resolution, sensitivity, time, cost, volumes of sample required; (2) clarify influence of various technical variables in specimen collection, handling, and storage; (3) lay groundwork through evaluation of technology platforms and specimen handling for future studies of circulating proteins (biomarkers) in health and disease. To this effect, standardized samples were distributed to 18 participating laboratories and an integrated analysis of resulting data was carried out (Omenn et al., 2005). An initial integration exercise resulted in 3,020 proteins identified with two or more peptides. Rigorous statistical analysis taking into account the length of coding regions in genes, and multiple hypothesis-testing techniques resulted in a reduced set of 889 proteins identified with a confidence level of at least 95% (States et al., 2006). This pilot phase, though modest in scale, is illustrative of the merits of an organized, collaborative effort around a well-defined study. A full-scale plasma proteome project (Table 1) has substantial merit given the wealth of biological information that would result and that would serve as a basis for assessment of alterations of the plasma proteome-associated with disease. Such a project would also represent an important vehicle to guide the development of proteotypic peptides and other resources such as affinity capture agents that are needed for high-throughput studies. Certainly, other projects of a biological nature may be entertained such as characterizing the secretome of defined cell populations. Success in the execution of an ambitious project such as a plasma proteome project would encourage the launching of other similar projects.
Projects to Systematically Characterize Proteome Alterations That Occur in Well-Defined Disease States
Despite remarkable advances in our understanding of the molecular basis of common diseases such as cancer, substantial gaps remain both in our understanding of disease pathogenesis and in the development of effective strategies for assessing risk of developing disease, for early diagnosis, and for personalized treatment. A proteomic approach to characterizing disease complements and may well overcome some of the limitations of other approaches, for example, for risk assessment and early detection. The dynamic nature of the proteome of a cell or a tissue, which reflect responses to the microenvironment and to various stimuli and reflect alterations in the genome, provides ample justification for proteome level investigations. The opportunities as well as the challenges facing disease proteomics are daunting; hence, the need for an organized effort around disease states. Contributions of proteomics include: (1) delineation of altered protein expression, not only at the whole-cell or tissue levels, but also in subcellular structures, in protein complexes and in biological fluids; (2) the development of novel biomarkers for risk assessment, early detection, and diagnosis of disease; and (3) the potential for accelerating drug development through more effective strategies to evaluate therapeutic effect and toxicity.
Of particular interest is the development of disease markers using proteomics. A PubMed search using the terms serum markers and proteomics yielded nearly 4,000 publications reflecting activity in this field, which encompasses most diseases. Although much remains to be discovered in the field of biomarkers, clearly proteomics technologies currently available hold substantial promise in identifying markers of risk for developing disease, markers for early detection and blood-based markers for classifying disease and monitoring response to therapy. We have applied the same in-depth quantitative proteomic platform utilized for assessment of hormone therapy response, to identify novel proteins associated with risk for CHD or stroke among postmenopausal women (Pitteri et al., 2010). A pooling strategy was applied to plasma samples from 800 women who subsequently developed CHD and 800 women who subsequently developed stroke during Women's Health Initiative cohort follow-up. Case versus control concentration differences were observed for 37 proteins (p < 0.05) for CHD, some of which had false discovery rates < 0.05. Corresponding numbers for stroke were 47 proteins with p < 0.05, three of which, apolipoprotein A-II precursor (APOA2), peptidyl-prolyl isomerase A (PPIA), and insulin-like growth factor binding protein 4 (IGFBP4), had false discovery rate < 0.05. Other proteins involved in insulin-like growth factor signaling were also highly ranked. The associations of B2M with CHD (p < 0.001) and IGFBP4 with stroke (p = 0.005) were confirmed using ELISA, and changes in these proteins following the initiation of hormone therapy use were shown to have potential to help explain hormone therapy effects on those diseases. These findings point to the merits of deep proteome interrogation for uncovering disease-related proteins in this cases uncovering protein changes before onset of disease. A cumulative analysis of proteins identified to date in various in-depth proteomic studies using serum or plasma that we have undertaken has yielded protein products for over 6,000 proteins (Fig. 3). Proteins identified in serum or plasma are not limited to secreted proteins, particularly for proteins that occur in the low abundance range (Fig. 4).
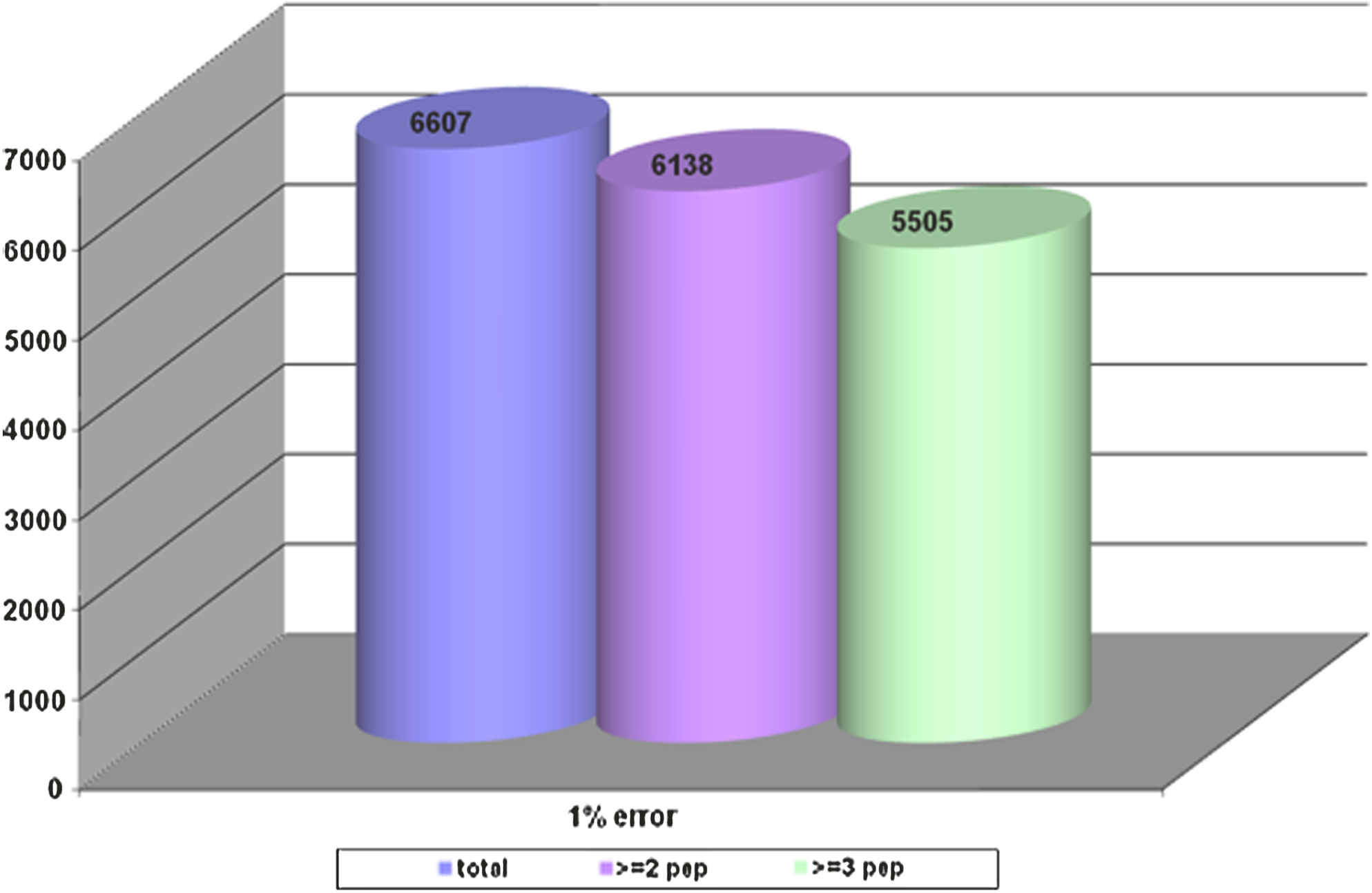
Cumulative numbers of unique plasma proteins identified by in-depth mass spectrometry based profiling listed according to numbers of corresponding peptides identified.

Distribution of proteins identified in plasma according to subcellular compartment.
Moving Forward with Collaborative Studies in Disease Proteomics
It is clear that current proteomics technologies allow in-depth analysis of major slices of the human proteome that have relevance to our understanding of disease and to the mining of the human proteome that impacts on disease prevention and treatment. However, given the complexities involved there remains a need for structuring large-scale ambitious proteomics projects as organized collaborative efforts. Such efforts may focus on a particular compartment of the proteome or a particular disease state. Emphasis will need to be placed on the procurement of high-quality specimens that avoid biases resulting from sample collection procedures and other confounding conditions. Particularly useful sources of specimens are those collected through clinical trials and through cohort studies as described for studies involving the large Women's Health Initiative cohort. It is essential for such collaborative projects to be milestone driven with resulting data deposited in repositories that allow the data to be readily interrogated and integrated with other data to address new questions. In addition, such projects would represent an important resource to guide the development of reagents to further interrogate the human proteome and apply findings from discovery studies to high-throughput strategies for screening, early detection, and diagnosis. Success in such collaborative studies will drive further developments in resources and technology creating further opportunities for proteomics.
Footnotes
Author Disclosure Statement
The author declares that no conflicting financial interests exist.