
Abstract
Abstract
TP53 is the most widely mutated gene across all cancer types. In head and neck cancer, approximately half of the tumors are found to contain TP53 mutations, which are correlated to an increased risk for locoregional recurrence and poor outcomes. In this study a mutational profiling of TP53 exons 5–8 was performed on tumor, peritumor and normal tissues from 57 HNSCC patients by direct sequencing of genomic DNA and cDNA. Cloning/sequencing in tumors carrying multiple TP53 mutations and semiquantitative SNaPShot mutation assay was performed in order to assess eventual allelic expression imbalances (AEI). We identified 24 out of 57 HNSCC patients (42%) carrying TP53 mutations and 5 patients carrying the R213R polymorphism. Cloning of the genomic DNA encompassing TP53 exons 5–8 from tumors with multiple TP53 mutations revealed that alleles carrying different types of TP53 mutations are present in these tumors. TP53 missense and nonsense mutations exhibit higher and lower TP53 transcript abundance compared to wild-type TP53 allele, respectively. Interestingly, three out of four patients with the R213R polymorphism analyzed were found positive for TP53 loss of heterozygosity (LOH) and also presented higher transcript abundance than the wild-type counterpart, specifically, in the tumor tissue and not in peritumor or normal tissues. HNSCC tumors present heterogenic cell populations carrying different TP53 mutations. All HNSCC samples analyzed show an alteration in the expression of mutated TP53 mRNA compared to the wild-type allele, most likely independently from the TP53 hemizygous status. The higher expression of R213R TP53 polymorphic allele in cancer tissue compared to normal tissue demonstrates a noninherited variation in allelic expression, independently from its mutation status for exons 5–8, suggesting a potential contribution to TP53 expression in HNSCC disease.
Introduction
The p53 tumor suppressor gene (TP53), is the most frequent target in genetic alterations in human cancers, with a prevalence of 20–60% in HNSCC (Edström et al., 2001; Nogueira et al., 1998). The region containing exons 5–8, encoding for the evolutionary conserved DNA-binding domain, represents the major site of TP53 mutations. After mutation in one TP53 allele, the remaining wild-type allele is often deleted and therefore, the mutant phenotype is expressed (Kiuru et al., 1997; Yin et al., 1993). Countless evidences has demonstrated that at least certain mutant forms of p53 protein may possess gain of function activity, thereby positively contributing to cancer progression. When the mutational analysis of TP53 and clinical outcomes in HNSCC were correlated, the results obtained were contradictory. A number of studies reported that TP53 gene mutations are associated with an increased risk of recurrences and poor outcomes (reviewed in Thomas et al., 2005). Alternatively, others showed that TP53 mutations or overexpression do not independently predict clinical outcomes in HNSCC (reviewed in Thomas et al., 2005). This discrepancy may probably be due to differences in techniques analysis. Apart from mutations numerous polymorphisms are also present at the TP53 locus (reviewed in Whibley et al., 2009). Among these polymorphisms, eight are exonic and synonymous. Although these SNPs do not change the structure of the protein, changes in base sequence could modify protein expression. A synonymous substitution at codon 36 was in fact shown to affect p53 function by influencing MDM2-mediated control of the translation of TP53 mRNA (Candeias et al., 2008). In addition, the recent discovery that microRNAs can inhibit translation by targeting the coding sequences of genes invites speculation that such a mechanism might also influence p53 expression control, making the study of TP53 sequence variants of crucial relevance. No exhaustive and accurate comparative studies for Allelic Expression Imbalances (AEI) of TP53 variants have been conducted so far in HNSCC.
In this study, we performed TP53 mutational analysis in tumor, peritumor, and normal tissues from 57 HNSCC patients. By cloning and sequencing TP53 cDNA from tumors carrying double or triple mutations, we found that different TP53 mutated alleles were present. The loss of heterozygosity (LOH) analysis showed that hemizygous TP53 mutations slight outweigh heterozygous mutations, suggesting that heterogenic cell populations compose these tumors. Semiquantitative SNaPshot analysis confirmed a positive AEI in favor of missense mutations in our HNSCC samples. Interestingly, we observed a higher expression of the synonymous R213R polymorphic allele compared to the wild-type allele, which is related to TP53 LOH positive status, but independently from its mutation status for 5–8 exons. This is specifically the case in cancer tissues and not in normal and peritumoral tissues, demonstrating a noninherited variation in allelic expression for this polymorphism.
Materials and Methods
Tissue samples preparation
Fifty-seven patients with primary HNSCC and any previous treatment with radiotherapy or chemotherapy who underwent resection in the Otolaryngology Head and Neck Surgery Department were included in this study. Three biopsies, from tumor, macroscopically free peritumoral tissue, and histologically normal tissue, were obtained for each patient. The peritumoral fresh tissue was taken at a distance at least of 1 cm from the tumor.
Genomic DNA and RNA extraction
DNA and RNA was coextracted using Trizol (Invitrogen, Carlsbad, CA) from snap-frozen tissue of tumor, peritumor, and normal biopsies. One microgram of RNA was reverse-transcribed into single-stranded cDNA by M-MLV reverse transcriptase (Invitrogen). Appropriate formalin-fixed, paraffin-embedded (FFPE) tissue blocks were selected for DNA extraction and multiple serial sections were stained with H&E. Under an operating microscope, tumor and normal adjacent epithelium of mucosa were dissected from the corresponding unstained 10-μm sections. These tissues were collected, deparaffinized, and followed by DNA isolation with the QIAamp DNA FFPE Tissue Kit (Qiagen, Chatsworth, CA).
PCR amplification of exons 5–8 of TP53 and mutational analysis by direct sequencing and SNaPshot assay
Primers were designed to amplify exons 5–8 of TP53 from DNA and cDNA as the template (Supplementary Table 1). The standard PCR was performed by using AmpliTaq Gold (Applied Biosystems, Bedford, MA) as shown in the Supplementary Data. PCR fragments obtained from the DNA and cDNA template were purified with NucleoSpin Extract II (Macherey-Nagel, Duren, Germany) kit and eluted with water. PCR products were sequenced directly by using the Big Dye V3.1 Cycle-Sequencing kit (Applied Biosystems) with M13 Fw and M13 Rw primers. Sequencing reactions were analyzed on to 3130 Genetic Analyzer (Applied Biosystems). A SNaPshot assay was performed for the TP53 codon mutations shown in Table 2 and in Supplementary Table 2. PCR products were analyzed for these mutations using the ABI PRISM SNaPshot Multiplex Kit (Applied Biosystems), according to the protocol supplied by the manufacturer. All primers were designed with a similar melting temperature and were checked for the absence of base pairing with other SNaPshot primers. The primer extension was conducted and amplified by 25 cycles of 96°C for 10 s, 58°C for 5 s, and 60°C for 30 s, followed by purification with Centrisep spin columns (Princeton Separations, Freehold, NJ). One to 2 μL of the reaction products were analyzed onto a 3130 Genetic Analyzer (Applied Biosystems).
LOH analysis
The LOH analysis was performed using paired tumor specimens and corresponding peritumor and normal tissues. The analysis was conducted on DNA obtained from fresh and FFPE tissue samples. Genomic DNA was analyzed for LOH by using three polymorphic markers for the TP53 locus (Supplementary Table 3). Forward primers were 5′ end fluorescence-labeled. PCR were performed in thermocycling conditions individually established for each pair of primers. For the characterization of alleles, PCR products were analyzed onto a 3130 Genetic Analyzer (Applied Biosystems).
Allele-specific gene expression measurement
Single nucleotide polymorphisms (SNPs) coding for missense and nonsense mutations and for the R213R polymorphism within the TP53 exons were selected as markers for allelic expression. All SNPs tested are listed in Supplementary Table 2. The PCR products specific for different TP53 exon portions were amplified from DNA and cDNA of fresh tumor tissue were purified as describe above. Primer extension reactions, purification, and electrophoresis analyses were carried out as just described above. The two alleles were distinguished by fluorescently labeled nucleotides of the SNP in both genomic DNA and cDNA, and the peak area was used to determine the ratio of the two alleles similar to the LOH analysis. The ratio of the two alleles of the cDNA from the transcript was normalized with the ratio from the genomic DNA as described (He et al., 2005).
Immunohistochemistry and HPV screening
Immunohistochemistry was performed using a standard avidin–biotin peroxidase method with monoclonal mouse antihuman p53 protein mAb clone DO-7 (1:25 dilution, Dako Ltd, Ely, UK) on 5-μm array sections after 20-min heat-induced epitope retrieval.
HPV was detected by the morphologic analysis on the tumor section stained with H&E seeking koilocytosis, koilocytotic atypia, or binucleation on the surface of squamous epithelium.
Results and Discussion
Fifty-seven patients with HNSCC, including larynx, pharynx, and oral cavity, entered the study. The characteristics of patients are presented in Table 1. We assessed the TP53 status by direct sequencing analysis of exons 5–8 on both the genomic DNA and cDNA obtained from fresh-frozen tumor, peritumor, and normal tissues for each patient. Twenty-four out of 57 HNSCC patients (42%) carried single or multiple TP53 mutations for a total of 28 mutations (Table 2 and Supplementary Table 4), according to the frequency of mutations reported for Italy in the IARC database (www-p53.iarc.fr). The majority of TP53 mutations are single base substitutions; some of them are missense (18/28), whereas others are nonsense (4/28) mutations. 6 out of 28 mutations are frameshift mutations caused by deletions and three of them lead to a predicted truncated p53 protein. Three tumors carried multiple TP53 mutations (V157I-R158H; del.T155-M169 and C242Y; R156H-C275S-R282Q). In addition, five patients carried the R213R synonymous polymorphism, among which two tumors carried the R213R polymorphism combined with a missense mutation (R213R plus Y234C) or a frameshift mutation (R213R plus a del.S106), respectively (Table 2). The TP53 status was also evaluated in the peritumoral and normal counterparts and a wild-type TP53 sequence was found in all cases except two (Supplementary Table 4). The same results were obtained by the mutational analysis on DNA and cDNA samples.
HNSCC, hand and neck squamous cell carcinoma.
The cells poorly differentiated overexpress p53 protein more than the tumor cells moderatately or well differentiated.
The overexpression of protein is not present in all tumor cells of the sample, but indipendently from the tumor cell differentiation.
Normal tissue shows a few cells with positive staining for p53 on the basal epithelium.
+p53 mutated allele is more expressed than wild-type allele; −p53 wild allele is more expressed than mutated allele; miss, missense; nons, nonsense; poly, polymorphism; AI, any information; NI, not informative (when at least two loci are homozygous); no AEI, absence of allelic expression imbalance between TP53 wild-type allele and polymorphic one.
To further verify the presence of mutations, we also performed semiquantitative SNaPshot assays on genomic DNA extracted from histological sections, in order to analyze cells whose origin from the tumor or peritumor areas was controlled at the microscopic level. The SNaPshot assay is indeed more sensitive than direct sequencing in the detection of mutations (Hurst et al., 2009). We analyzed FFPE tumor and peritumor tissues, obtained by manual microdissection, of seven HNSCCs carrying mutated TP53. All the analyzed patients showed mutations in the tumor but not in the adjacent normal tissue (Fig. 1).

TP53 sequencing analysis in tumor and adjacent normal tissue of HNSCC patients. The SNaPshot analysis for the TP53 R175H substitution performed on genomic DNA obtained from tumor (
By cloning the exons 5–8 from TP53 cDNA, we subsequently evaluated the allelic localization of the above-mentioned double and triple TP53 mutations. The sequencing analysis of about 15 clones for every TP53 fragment showed the allelic distribution. The triple TP53 mutant, R156H-C275S-R282Q, presented the mutations R156H-C275S and R282Q on independent alleles (Supplementary Fig. 1). Also, the double TP53 mutant del.T155-M169 and C242Y presented the two mutations on different alleles. The double mutant V157I-R158H presented two different mutant allelic populations: one with the R158H mutant alone and the second with both TP53 mutations V157I and R158H, suggesting that the V157I mutation occurred later than R158H. Finally, in the patient carrying TP53 R213R-Y234C, the polymorphism and the mutation were localized on the same allele. As shown in Table 2, the LOH analysis showed a slight predominance of hemizygous TP53 mutations compared to heterozygous mutations (11 out of 18 informative cases). These data reinforce the previously proposed concept (Wang et al., 2006) that heterogenic clonal cell populations are present in HNSCC.
In order to analyze the AEI of missense, nonsense, and frameshift TP53 mutations a SNaPshot semiquantitative mutational assay was applied to a subset of 17 mutated TP53-carrying tumors (Table 2). In this assay, the ratio between the amount of mutated (or polymorphic) and wild-type alleles obtained from the cDNA is normalized over those obtained from the genomic DNA, producing the allelic expression ratio (see Materials and Methods), that represents the abundance of the variant allele over the wild-type (Fig. 2). The allelic expression ratios of missense mutations (G266R, R248Q, R175H, P278T, C242Y, Y205C, K132N, H193R, Y234C, R156H, C275S, R282Q) were ≥1.5, indicating that the abundance of missense mutated alleles was at least 33.3% higher than that of the wild type (Fig. 2a and d and Table 2). The allelic expression ratios of the three nonsense mutations analyzed (W146X, E198X, C277X) were ≤0.67, indicating that the expression of nonsense mutated alleles was lower than the wild-type allele (Fig. 2c and Table 2). The frameshift mutation (del.M243), which leads to a predicted truncated p53 protein, showed a decreased mRNA abundance, as expected (Fig. 2b and Table 2). These results show a significant difference in the mRNA abundance among the different mutational forms, possibly independently from the hemizygous or heterozygous status of TP53 (Table 2).
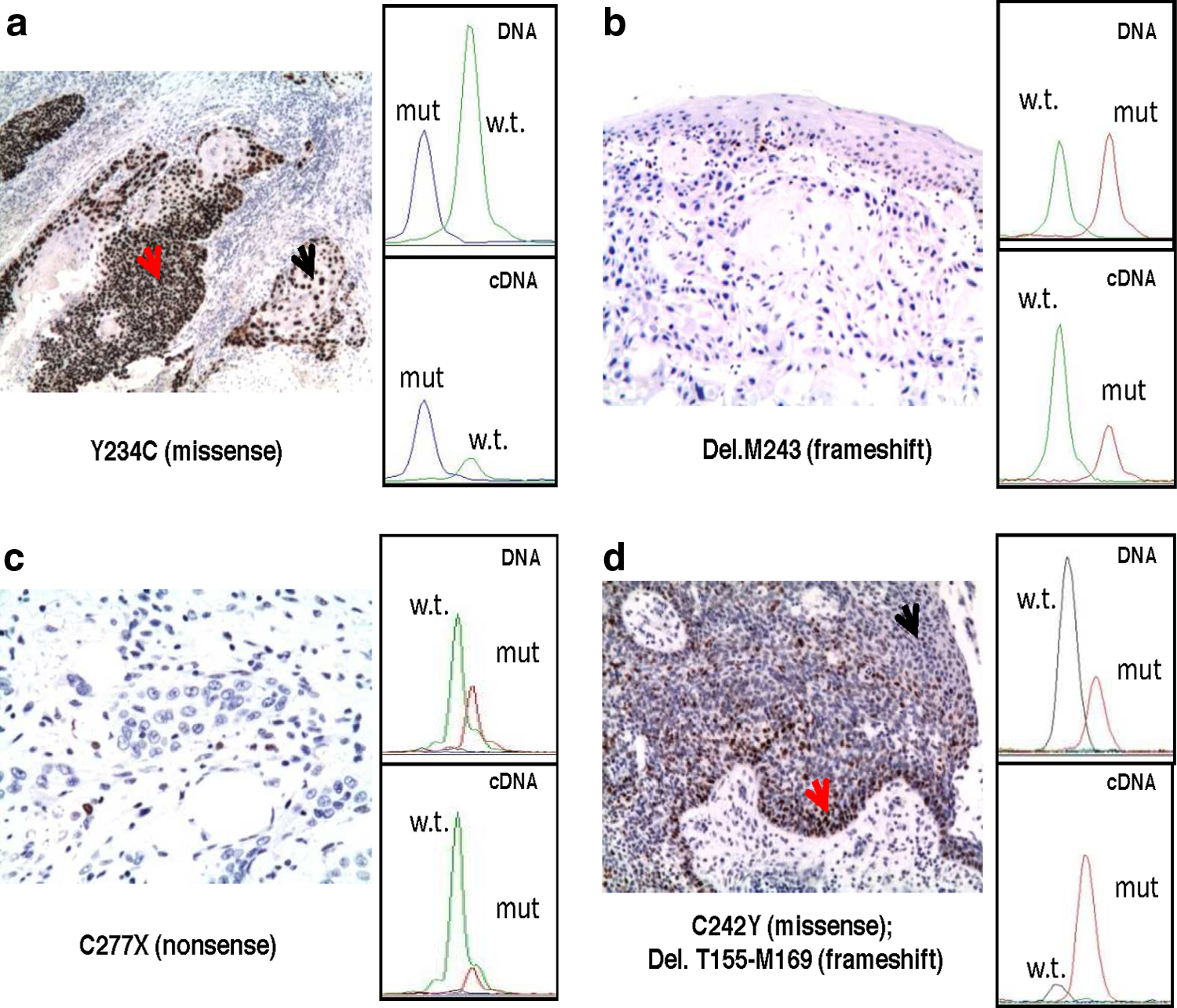
Correlation between AEI of missense, nonsense, and frameshift TP53 mutations and p53 protein expression in HNSCC tissues. p53 protein expression, evaluated by immunohistochemistry (IHC), and the relative AEI, evaluated by SNaPshot analysis, from four tissues representing different TP53 mutation types are shown. (
Next, we performed the SNaPshot assay on tumor, peritumor, and normal tissues from four out five TP53 R213R-carrying patients. The semiquantitative primer extension assay showed a higher R213R allelic expression in cancer tissue in comparison to normal tissue, suggesting a noninherited variation in allelic expression for this polymorphism compared to the wild-type allele (Figs. 3 and 4 and Table 2). The higher expression of R213R allele was related to the positive TP53 LOH status, but independently from its mutation status for 5–8 exons (Fig. 4 and Table 2).

Correlation between AEI of R213R TP53 polymorphism and p53 protein expression in normal, peritumor, and tumor tissues. p53 protein expression, evaluated by IHC, and the relative AEI, evaluated by SNaPshot analysis, from patient 58, carrying R213R polymorphism, are shown. TP53 R213R allele shows higher expression of both mRNA and protein specifically in the tumor tissue (
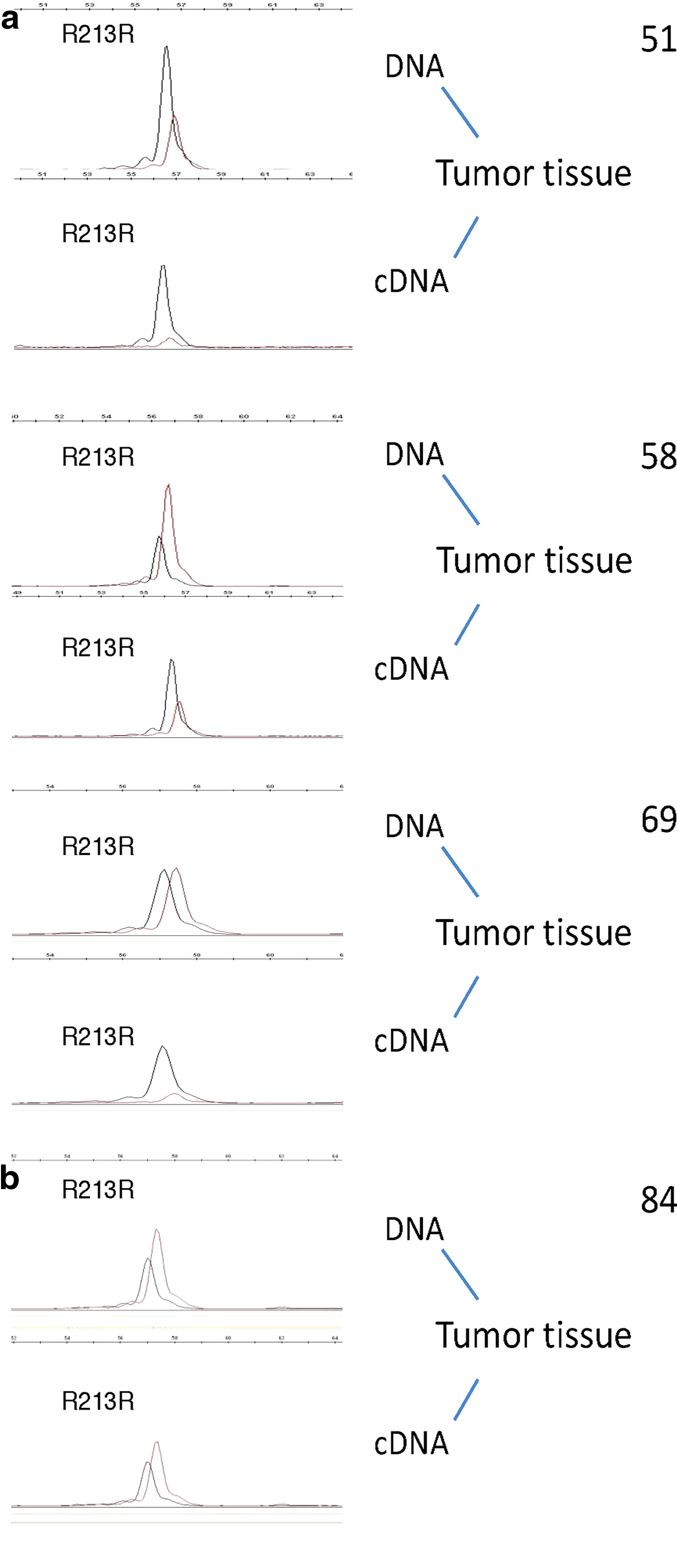
Comparison of p53 AEI among four patients carrying the R213R polymorphism. SNaPshot analysis of R213R expression was performed on cDNAs from the tumor tissues of the indicated patients. (
To evaluate the correlation level between AEI and p53 protein expression in patients with TP53 sequence variants, we analyzed p53 protein by immunohistochemistry (IHC) in 24 HNSCC (Fig. 2 and Table 2). Squamous epithelia obtained from the normal mucosa of these 24 patients never reacted positively to the p53 antibody, except one sample carrying the TP53 R213R polymorphism, which presented about 30% of positivity. A few normal tissues showed some p53 positive cells in the basal epithelium. Thirteen of 14 HNSCC specimens with TP53 missense mutations were found positive for p53 protein overexpression (Table 2). The only sample with TP53 missense mutation (patient 29) negative for IHC was positive for HPV detection. Interestingly, in many cases we observed differential p53 immunoreactivity distribution into the same tumor sample, with poorly differentiated areas expressing higher levels of p53 protein than moderately or well-differentiated areas. As expected, all four HNSCC with nonsense mutations were found negative for p53 expression (Fig. 2c and Table 2). Both AEI and IHC results are available for 16 HNSCCs with TP53 mutations and/or R231R polymorphism and HPV status. These data showed a perfect correlation between allelic expression of missense, nonsense, and frameshift mutations and p53 protein expression (Table 2).
Conclusions
TP53 missense and nonsense mutations have a dissimilar AEI behavior in HNSCC, most likely independently from the LOH TP53 status. The higher expression of missense mutant alleles could be caused by the inactivation of the wild-type allele, as well as by the higher expression or mRNA stability of mutated alleles. Comparatively, to the nonsense mutations, we hypothesize that their low expression may be due to the nonsense mediated mRNA decay (NMD) surveillance pathway, which ensures the rapid degradation of the majority of mRNAs containing premature translation termination codons (Mühlemann et al., 2008).
A previous genetic study showed that R213R does not represent a genetic susceptibility marker for the development of gastroesophageal reflux disease to Barrett's esophagus and esophageal cancer in a group of Brazilian patients (Pilger et al., 2007). Our data suggest that the R213R polymorphism could take part in mRNA overexpression of TP53 in HNSCC. Interestingly, the five R213R patients are all heterozygous for this locus and the higher expression of R213R allele was related to TP53 LOH positive status but independently from its mutation status for 5–8 exons. It may be possible that the two R213R patients with wild-type TP53 forms (TP53 LOH positive) carry-on other mutations spanning on different exons that may favor the specific-polymorphic allele expression. In addition, we were able to show that the R213R mRNA is more abundant when a genetic instability in HNSCC cells is present. Further studies will shed light on the significance of the expression of this and other allelic variations for HNSCC tumor progression, prediction of treatments response, and survival.
Footnotes
Acknowledgments
The authors thank Mrs. Annarita Pennetti and Mrs. Arianna Papadantonakis for their skilfull and first rate technical assistance. We greatly appreciate the support given by the AIRC-ROC to the oncogenomic platform and the AIRC to G.B. and S.S.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.