
Abstract
Abstract
The histological grade/stage of tumor is widely acknowledged as an important clinical prognostic factor for cancer progression. Recent experimental studies have explored the following two topics at the molecular level: (1) whether or not gene expression levels vary by different degrees among different tumor grades/stages, and (2) whether some well-defined modules could distinguish one grade/stage from another. In this article, using breast cancer as an example, we investigated this topic and identified grade/stage-related active modules under the framework of a weighted network integrated from a human protein interaction network and a transcriptional regulatory network. Our results enabled us to draw the conclusion that the gene expression profile could provide more clues about tumor grade, but reveals less evidence about tumor stage. In addition, we found that our modular biomarker method had additional advantages in identifying some tumor grade/stage-related genes with slightly altered expression. According to our case study, the framework we introduced could be used for other cancers to identify their modules during grading or staging.
Introduction
With the development of high-throughput methods over the last decade, considerable efforts and financial resources have been focused on identifying molecular biomarkers for cancers by correlating gene expression patterns with disease phenotypes of interest (Perou et al., 2000; van't Veer et al., 2002; Wang et al., 2005). Hempel and associates and Kempkensteffen and colleagues reported that genes with significantly altered expression levels were able to distinguish different tumor stages or grades based on gene expression analyses (Hempel et al., 2009; Kempkensteffen et al., 2007). However, a strict single-gene model might not only overlook some tumor grade/stage-related genes with slightly altered expression levels, but it may also miss valuable information such as the sophisticated interactions and regulation relationships among genes (Ransohoff, 2004; Simon et al., 2003). An increasing number of cancer studies have combined human gene expression profiles with computational-based module searching algorithms to obtain a more comprehensive view of the molecular underpinning of cancers (Colak et al., 2010; Li et al., 2010; Segal et al., 2005). Genes within a module appear to have similar expression patterns, share common underlying regulatory mechanisms, and thus have strong associations with specific biological functions that determine the behavior or phenotype of the cell (Michalak, 2008; Purmann et al., 2007). These module-based approaches are aimed at a more robust and interpretable characterization of the dynamic transcriptional changes seen during the development of cancers (Wang et al., 2008). Under the framework of an integrative network, a module-based biomarker identification method could enable researchers to identify active modules to elucidate their possible roles in tumor staging or grading when coupled with gene expression profiles.
In this article, using breast cancer as an example, we introduced an integrated method to identify tumor grade/stage-related active modules under the framework of a human integrative network. First, co-regulated modules were detected with Co-Regulatory Analysis using Integrated Networks (CRAIN) in an integrative network from a human protein interaction network and a transcriptional regulatory network. Then according to the expression profile of breast cancer, the activity scores and their corresponding significant levels were calculated for these modules to screen tumor-related active modules. At the next step, the Jonckheere-Terpstra test was introduced to identify tumor grade/stage-related active modules in a grade/stage-related expression profile. Finally, further functional annotations and literature retrievals were used to decipher and evaluate their detailed roles in tumor staging and grading progression.
Materials and Methods
Human interaction data sources
The human protein-protein interaction data were extracted from the Human Protein Reference Database (HPRD) (Peri et al., 2004). The derived interaction network contained 34,083 interactions between 9014 proteins.
The transcriptional regulatory data was acquired from the Transfac Database (release 11.4) (Matys et al., 2003). The resulting regulatory network consisted of 281 transcription factors and 624 genes with 1603 interactions. For further analysis, we assigned an empirical weight to be 0.99 (a balanced confidence level of each edge) for each transcriptional regulatory interaction.
Breast cancer gene expression data
Breast cancer gene expression datasets were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The accession number of the first dataset was GSE5364 from platform GPL96 (including 183 breast cancer samples and 13 normal samples). The accession number of the second dataset (with breast cancer stage and grade) was GSE2109 from platform GPL570 (Table 1).
The method of breast tumor grading used was the Nottingham Grading System. It stratified breast tumors into grade 1 (G1; well-differentiated, slow-growing), grade 2 (G2; moderately differentiated), and grade 3 (G3; poorly differentiated, highly proliferative) malignancies.
The method of breast tumor staging used was the TNM staging system. We could determine the development of the tumor by the size of the tumor, and whether the tumor was invading nearby tissues and spreading to other parts of the body.
Detection of co-regulated modules
We scaled and merged a human protein-protein interaction network and a transcriptional regulatory network to construct an integrated weighted network. Then human co-regulated modules were detected from the integrative network using a probabilistic model (CRAIN) (Chen et al., 2010).
Gene expression significance analysis
We first measured the significance of differential expression of each gene. Here we used the Student's t-test to extract the significance of the change of a gene. The obtained p value (pi) of each gene (gene i) was converted to a z-score (zi) by using the inverse normal cumulative distribution function (θ−1). The z-scores followed a standard normal distribution, and a higher z-score indicated a more significantly induced gene expression.
Activity score calculation of co-regulated modules
The activity score represented the altered form of the co-regulated modules. Expression levels of significantly changed genes and transcription factors in a module were selectively observed to measure the activity score of the module. First, a set of z-scores (z (pj) ) for genes and transcription factors in module j was assembled. Then the set of z-scores in module j was sorted in descending order (z′ (pj) ). By expanding from the maximum z-score, top t z-scores that maximized the activity score were selected. The activity score of a module (A (pj) ) was an aggregated form of the expression scores of genes (z′ji) in a module j that maximized the aggregated score.
where j was the module j, k was the number of genes in module j, and t was the number of genes of which z-scores maximized the activity score.
Active modules screening
The screening of active modules was accomplished by normalization of each A(pj) score to an Acorrected (pj) score, which was executed by correcting for background distribution based on the mean (μ
k
) and standard deviation (σ
k
) as derived from A(pj) scores of 10,000 random sets of k genes. Among these, the 10,000 random gene sets of each module were simulated by randomly selecting genes from background gene sets with equal numbers of module genes. The basic implication of the scoring function for identifying active modules was a statistical test for the null hypothesis: “observed gene expression of genes and transcription factors of a module were purely random.”
Once we obtained corrected activity scores, p values were calculated using the inverse normal cumulative distribution function (θ−1). A p value of 0.05 was used to select active modules.
Grade/stage-related active module identification
The Jonckheere-Terpstra test was a useful variation in the analysis of ordered categorical data (http://www.mathworks.com/matlabcentral/fileexchange/22159-jonckheere-terpstra-test-on-trend/content/jttrend.m). We used it to evaluate the correlation between the expression level of each active module and breast tumor stage or grade. The null hypothesis of the test was that there were no differences among tumor grades/stages. The alternative hypothesis was that the sample means changed along the ordered sequence of tumor grades/stages. The test was appropriate and powerful due to the reliance on fewer assumptions. It was also intuitive and simple to carry out by hand, at least for our small samples. Average expression of all genes in a module was used to represent the overall expression level of the module.
Results and Discussion
The breast cancer-related active modules found
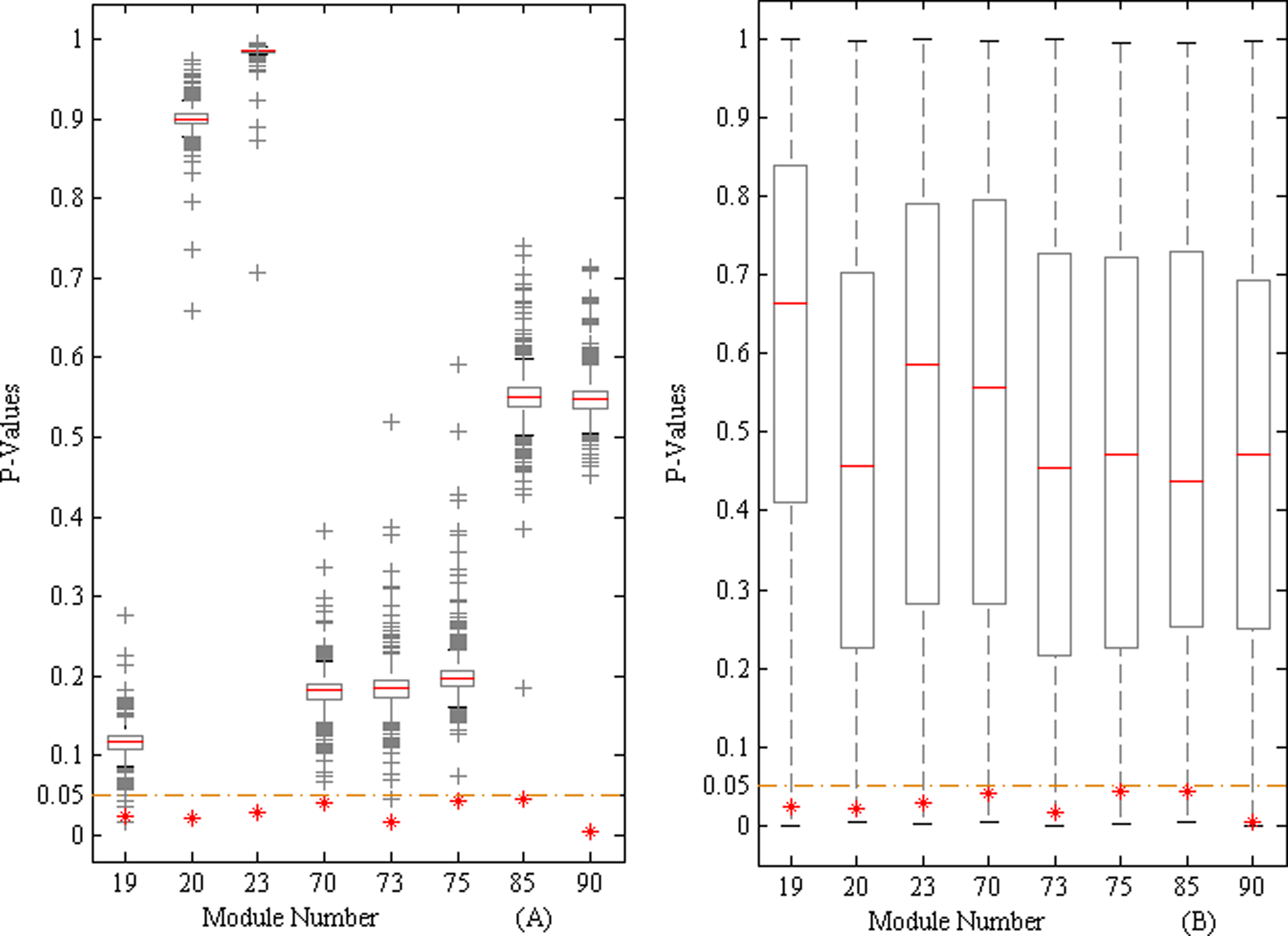
With several scoring and computational steps (Fig. 1), we identified eight active modules related to breast cancer from 96 human co-regulated modules, which were detected by the integrative weighted network. Each module contained one or more transcription factors (Fig. 2). To enhance technical clarity of the method and demonstrate the stability and reliability of the active modules, we performed several simulation studies in view of its sensitivity to error structure and error distribution. First, we conducted random replacements for the nodes of each module with an equal number of proteins/transcription factors (TFs) from the background integrative network. This process kept the degree of module nodes constant. For each of the 96 human co-regulated modules, we repeated this random replacement 1000 times (Supplementary Fig. S1; see online supplementary material at http://www.liebertonline.com). Second, we randomly selected tumor and normal samples with the sample label perturbation from the background gene expression profile GSE5364. This simulation process was repeated 1000 times. Then active module screening was performed with the 1000 random gene expression profiles (Supplementary Fig. S2; see online supplementary material at http://www.liebertonline.com). It is worth noting that the eight active modules that we screened were significant in random tests of frequency <5% for both kinds of simulation studies (Fig. 3), suggesting that the active modules were authentic.

Method overview. (
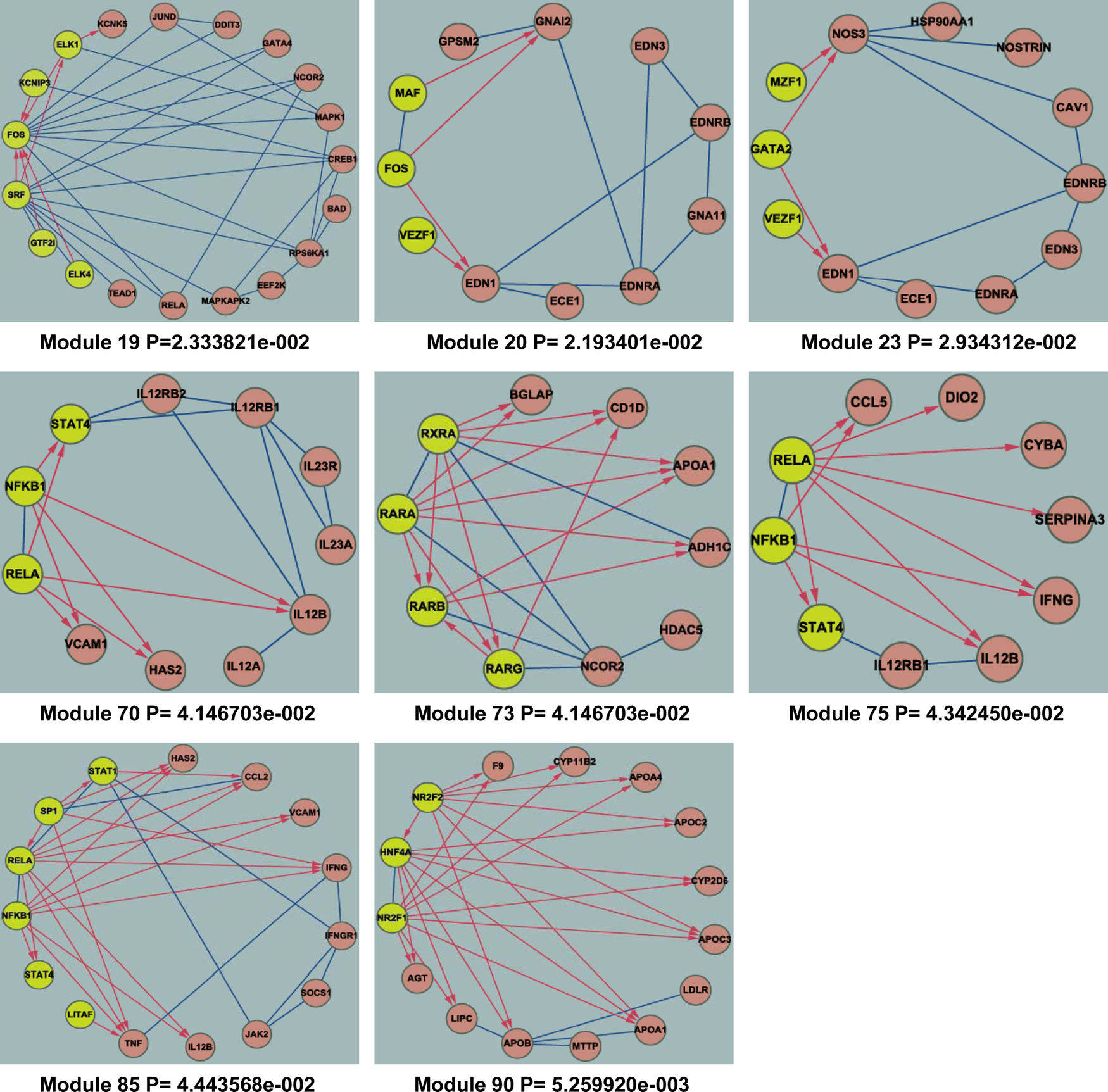
Eight active modules. After several scoring and computational steps, eight active modules (modules 19, 20, 23, 70, 73, 75, 85, and 90) were identified to be related with breast cancer. The figure shows these eight active modules and their significant levels. For each module, yellow nodes represent TFs and red nodes represent proteins. Interactions between TFs and proteins are red; interactions between protein pairs are blue.
Simulation study for the eight active modules. (
Gene ontology (GO) enrichment analysis and KEGG annotation analysis were performed for each active module using DAVID (count ≥2; EASE ≤0.05) (Dennis et al., 2003; Huang da et al., 2009), a useful tool that integrates different sources of biological information to obtain biological annotations. The results showed that each module had significantly annotated breast cancer-related GO information (Supplementary Table S1; see online supplementary material at http://www.liebertonline.com), such as mesenchymal cell differentiation (modules 20 and 23) (Molloy et al., 2009), positive regulation of cell proliferation (module 70) (Mester and Redeuilh, 2008), regulation of apoptosis (Vegran et al., 2011), and regulation of programmed cell death (module 85) (Schlotter et al., 2008). According to KEGG annotation, co-regulated modules could organize multiple biological pathways (Supplementary Table S1; see online supplementary material at http://www.liebertonline.com). Some of them tend to be annotated in the same pathways, such as pathways in cancer (modules 19 and 73), and the Jak-STAT signal pathway (modules 70 and 85). Studies have showed that aberrant activation of the Jak-STAT pathway might predispose to malignancy by mediating the signals of a wide range of cytokines, growth factors, and hormones due to deregulation of proliferation, differentiation, or apoptosis (Hernandez-Vargas et al., 2011; Vaclavicek et al., 2007). We noted that the modules had similar functions, such as cell differentiation or cell development. Thus it was imperative to seek more valuable information about these active modules.
Tumor grade/stage-related features of expression-altered active modules
Expression-altered modules provided clues of tumor grade
The pathological grade was related to the degree of tumor differentiation, and was an index of the development of malignant disease (Ignatiadis and Sotiriou, 2008). We evaluated the correlation between the expression level of each active module and breast tumor grade using the Jonckheere-Terpstra test. The results showed that seven out of eight active modules were significantly correlated with breast tumor grade (p<0.01; Table 2). We randomly selected different grade samples with sample label perturbation from the background gene expression profile GSE2109. We repeated this random process 1000 times, and found that for all of the seven grade-related active modules that we screened, random tests of frequency <5% could achieve significant results (Fig. 4), suggesting that the grade-related active modules were reliable.

Simulation study for the seven grade-related active modules. Samples of different grades were randomly selected 1000 times with the sample label perturbation from the background gene expression profile GSE2109. Random tests of frequency <5% achieved significant results for each grade-related active module. The box plot shows the results of random testing. The x-axis shows the module number of seven grade-related active modules, and the y-axis the significant levels. Red stars represent the true significance levels of grade-related active modules. The yellow dashed line represents the threshold value of screening grade-related active modules.
The table lists the significance levels of tumor grading for eight active modules.
NS, non-significant.
Our active co-regulated modules showed a good ability to distinguish different breast tumor grades, which was consistent with a recent long-term follow-up study from the Nottingham group (Rakha et al., 2008). This could be the result of a co-regulated mechanism intervening. Each module assembled a set of genes with similar biological functions that were regulated by one or more TFs. For example, module 70 consisted of three TFs (NFKB1, RELA, and STAT4), and eight genes (Fig. 2). NFKB1 and RELA, the NF-κB genes, were closely related to breast cancer cell proliferation, invasiveness, angiogenesis, and inflammation, and might serve as novel therapeutic targets (Lerebours et al., 2008; Zhang et al., 2007). Liu and associates reported that RELA was typically associated with transcriptional activation (Liu et al., 2011), which was essential for the activity of its target genes (such as HAS2). Expression of HAS2 impaired intercellular adhesion machinery and elicited cell survival signals in tumor cells (Koyama et al., 2007). Some studies have implicated a central role of HAS2 in the initiation and progression of breast cancer (Udabage et al., 2005). Another group (module 73) contained four TFs (RARA, RARG, RXRA, and RARB), and their six target genes (Fig. 2). Recent studies have shown that these TFs were important in carcinogenesis. In most breast carcinomas and breast cancer cell lines RARB was lost or downregulated, whereas RARA, RARG, and RXRA were variably expressed. This series of unexpected variations could have effects on tumor emergence, promotion, and metastasis (Peng et al., 2004). Not only RARB, but also RARA, RARG, and RXRA might serve as potential targets in breast cancer prevention and therapy trials (Klaholz et al., 2000). Their target genes, such as APOA1, the major apoprotein constituent of high-density lipoprotein, also play a major role in the invasion and metastasis of breast cancer (Hamrita et al., 2010; Han et al., 2008). Metastasis is the main feature of tumor grade, and indicates the malignant spread of the tumor. These findings demonstrate that one or more TFs in co-regulated modules may coordinate different genes with specific functions. Our active co-regulated modules regulated specific biological functions associated with tumor grade, thus contributing to the pathogenesis of disease. However, module 75 had too few protein-protein interactions, and nearly all regulation relationships were induced by only one transcription factor. This may be the reason module 75 could not distinguish tumor grade.
Gene expression profiles revealed little about tumor stage
The tumor stage was based on the size of the tumor, and whether the tumor was invading nearby tissues or spreading to other parts of the body. Thus the tumor stage reflected the extent of injury (Jeruss et al., 2008). We evaluated the correlation between the expression level of each active module and breast tumor stage by the Jonckheere-Terpstra test. There was one module (module 20) that correlated with breast tumor stage (p=0.0046) among the eight active modules.
Research findings have shown that significant global alterations in gene expression occur at the earliest stage of progression, and that these alterations are maintained in the later stages compared with patient-matched normal samples (Allred et al., 2001; Ma et al., 2003). Van't veer and colleagues also reported that breast cancer metastatic potential could be reliably predicted by the gene expression profile of the early stage of the primary tumor (Van't Veer et al., 2002). Our active modules showed only a weak ability to distinguish breast tumor stage, which was consistent with previous studies. The results implied that before developing the end-stage tumor, the gene expression profile of early-stage disease may reflect the progressive potential of the pathological lesion.
Slightly-altered genes were identified for tumor grade
At the same time, we evaluated the correlation between the expression level of each gene of the seven grade-related active modules and breast tumor grade. Aside from duplicate records, there were 78 genes in the seven active modules. Most of the genes did not show good grade-related distinguishing performance (Supplementary Table S2; see online supplementary material at http://www.liebertonline.com). These findings suggested that active module-based analyses could implicate genes with low discriminative potential (e.g., those that were only slightly altered), if such proteins participated in a module whose overall activity was discriminative (Chuang et al., 2007). Such genes could arise within an active module if they were essential for maintaining the integrity of the module. This property was important for the discovery of disease-causing genes. On the other hand, as far as we knew, some of the significantly-expressed genes in breast cancer grade were not known as breast cancer mutated genes. This further proved that this active module-based analysis could detect some important breast tumor grade-related genes that were not easily perceived before.
Performance evaluation
Though the factors that influenced the performance of an approach were various, we demonstrated that an active module-based approach was valuable. When evaluating the correlation between the expression level of each active module and breast tumor stage, we noted that the number of samples for stage 0 and stage 4 were relatively small. In order to assess whether the non-significant results of staging were due to the different amounts of samples for the different stages, we performed the Jonckheere-Terpstra test on samples of stage 1, stage 2, and stage 3, and excluded this influence from the results.
We also considered the influence of sample size when evaluating the correlation between the expression level of each active module and breast tumor grade. We randomly chose 30, 30, and 30 samples, respectively, from 31,112 and 140 samples for grade 1, grade 2, and grade 3, and repeated this random sampling 100 times. We also randomly chose 30, 60, and 60 samples, and 30, 90, and 90 samples, for grade 1, grade 2, and grade 3, respectively. With these random samples, we evaluated the correlation between the expression level of each active module and breast tumor grade by Jonckheere-Terpstra testing. The results showed that our grading results did not change with the sample size of each grade (Table 3). Thus we could conclude that our method was not dependent on sample size.
Samples that were randomly chosen respectively from 31,112 and 140 samples for grade 1, grade 2, and grade 3.
Samples that were randomly chosen respectively from 31,112 and 140 samples for grade 1, grade 2, and grade 3.
Samples that were randomly chosen respectively from 31,112 and 140 samples for grade 1, grade 2, and grade 3.
The table lists the number of screened significant grade-related active modules among 100 random samples.
In addition, by performing Jonckheere-Terpstra testing on the other breast tumor grade-related dataset GSE5460 (with 27 samples for grade 1, 32 samples for grade 2, and 70 samples for grade 3), we found that five of the eight active modules were related to breast tumor grade (Table 2). It is worth noting that the five modules were all part of our seven grade-related active modules. Because of the deficiency of the genes found in GSE5460, the genes in module 23 and module 70 did not exactly match the dataset, which led to two modules being missed.
Gene-based analysis
We know that 26 of 78 genes in the seven grade-related active modules were significantly related to breast tumor grade (Supplementary Table S2; see online supplementary material at http://www.liebertonline.com). We also evaluated the correlation between the expression level of each single gene of the grade-related gene set GSE2109 and breast tumor grade. Then we ranked all genes with significant results, and collected the first 100 and 300 genes, respectively. Only 3 and 7 of those were also seen in 26 significant genes of the seven grade-related active modules. These results proved that this active module-based analysis could detect more breast tumor grade-related genes that were not detected by gene-based analysis. Thus compared to gene-based analysis, our method could enable us to elucidate more grade-related information when coupled with gene expression profiles.
Finally we assembled a list of breast cancer grade-related susceptibility genes (Supplementary Table S3; see online supplementary material at http://www.liebertonline.com) that had been reported as such in previous research (Baskin and Yigitbasi, 2010; Choi et al., 2010; Kidokoro et al., 2008). Breast cancer susceptibility genes were on the lists of genes that harbored mutations causing breast cancer, which related to all aspects of breast cancer progression. Grade was one of the evaluation standards used for detecting whether or not a gene was a susceptibility gene, and was an index of the degree of tumor differentiation and the development of malignant disease. Though these susceptibility genes were not acquired by our active modules, we also found that none were significantly correlated with breast tumor grade by gene-based detection (Supplementary Table S3; see online supplementary material at http://www.liebertonline.com). Until now, most susceptibility genes were found by experimental approaches but not expression-based computational methods, so these susceptibility genes were not suitable for our expression-altered computational approach. Alternatively, our expression-altered computational approach could detect other important tumor grade-related genes, which supplement traditional experimental approaches.
Conclusions
In this article, a set of active co-regulated modules was identified for breast cancer by our analysis under the framework of a weighted network integrated from a human protein interaction network and a transcriptional regulatory network. According to the results of functional evaluations and a literature search, our expression-altered co-regulated modules showed good ability to distinguish breast tumor grade. These findings support a molecular basis for researching grade-related processes in breast cancer by means of gene expression profiles. Regarding tumor stage, gene expression profiles may yield fewer essential clues to carcinoma pathogenesis. We suggest that other evidence is needed to help assess tumor stage.
Furthermore, seven modules were found to be related to breast tumor grade, and 33.3% of all the genes in these modules were significantly associated with tumor grade. This result was consistent with previous single gene-based analyses. However, other genes appeared to have slightly-altered expression levels and showed no significant associations with tumor grade. This suggested that modular biomarkers could succeed in identifying slightly-altered genes associated with tumor grade, and that their associations might be largely due to their interconnection with significantly-expressed genes within each functional module. It should also be noted that further studies should be made of these slightly-altered tumor grade-related genes based on our module-based method. With future experiments providing more complete knowledge of the transcriptional regulation and interaction network, the analysis of co-regulatory mechanisms will allow researchers to identify other tumor-grade related modules.
Footnotes
Acknowledgments
This work was supported in part by the Science & Technology Research Project of the Heilongjiang Ministry of Education (grant no. 12511271), the National Natural Science Foundation of China (grant no. 61272388), and the Student Innovation Funds of Heilongjiang Province (grant no. 2012-011HLJ).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.