
Abstract
Abstract
Scoparone is an active ingredient of Yinchenhao (Artemisia annua L.), a well-known Chinese medicinal plant, and has been utilized in prevention and therapy of liver damage. However, the molecular drug targets associated with the pharmacological effects of scoparone are largely unknown. In the present article, we extend the previous research on Yinchenhao through a study of its active ingredient and thus the putative targets of scoparone. We employed two-dimensional gel electrophoresis, and all proteins expressed were identified by MALDI-TOF/TOF MS and database research. Protein-interacting networks and pathways were also mapped and evaluated. The possible protein network associated with scoparone was constructed, and contribution of these proteins to the protective effect of scoparone against the carbon tetrachloride-induced acute liver injury in rats are discussed herein. Hepatoprotective effects of scoparone on liver injury in rats were associated with regulated expression of six proteins which were closely related in our protein–protein interaction network, and appear to be involved in antioxidation and signal transduction, energy production, immunity, metabolism, and chaperoning. These observations collectively provide new insights on the molecular mechanisms of scoparone action against hepatic damage in rats.
Introduction
Identification of the putative targets of natural products is an important aspect of current drug discovery and will aid drug development (Gersch et al., 2012). Yinchenhao (Artemisia annua L.) is one of the most popular traditional Chinese medicinal plants for treatment of liver disorders (Sun et al., 2013; Wang et al., 2013; Zhang et al., 2011). It can be used clinically to treat liver fibrosis, cholestasis, hepatitis C, and cholestatic diseases (Yin et al., 2012). Interestingly, scoparone (Fig. 1), one of the main constituents of Yinchenhao, shows hepatoprotectivity and has been proven effective for liver injury (Wang et al., 2011; Zhang et al., 2012). Furthermore, scoparone was reported to be an effective drug to enhancing secretion of bile acids for normalizing liver function (Yang et al., 2011). It suggested that scoparone may be a good lead compound for further studies.

Chemical structure of scoparone.
Among the reported pharmacological properties of scoparone, its hepatoprotective effects are of particular interest. However, the molecular drug targets associated with the pharmacological effects of scoparone are largely unknown. Proteomic approaches could provide a recent breakthrough for the clarification of target proteins. Here, we will use proteomics approaches to explore the mechanisms and implications and molecular drug targets of scoparone. Moreover, a computational program, STRING (Jensen et al., 209), was applied to verify the drug targets of scoparone and to mine the functional association of the defined proteins.
Materials and Methods
Reagents
Ultra-pure reagents for polyacrylamide gel preparation were obtained from Bio-Rad. Dithiothreitol (DTT), TPCK-Trypsin, iodoacetamide, thiourea, and α-cyano-4-hydroxycinnaic acid were provided by Sigma Chemical (St. Louis, MO, USA). IPG buffer and 18-cm immobilized pH gradient (IPG) strips (pH 3–10) were purchased from Amersham Pharmacia Biotech (Uppsala, Sweden). All other reagents were purchased from Bio-Rad.
Animal sampling and handling
Animals were randomly assigned to scoparone, control, and model groups of 8 rats per group. The rats in the animal model group were orally administrated 20% CCl4 (1 ml/kg body weight) and scoparone for 8 consecutive days (at 8:00
Proteomics analysis
Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) was performed. Protein spots with scores that were higher than 61 were considered to be differentially expressed, and were selected and subjected for further identification by MALDI-TOF/TOF MS analysis. Two-dimensional gel electrophoresis, image analysis, protein identification, and database searching were performed. Detailed procedures are described in our previously published work that used the identical methodology (Sun et al., 2013).
Bioinformatics analysis of proteomic data
Identified proteins were further analyzed using STRING for protein–protein interactions, to statistically determine the functions and pathways most strongly associated with the protein list. Prior to upload and analysis using STRING, the mean ratio of each quantified protein in a group was calculated and the fold change between the groups calculated. The KEGG (www.genome.jp/kegg/) pathway database were used to further examine the significantly expressed molecules.
Results
Two-dimensional gel electrophoresis
Proteins from the control, model, and scoparone groups were analyzed by 2DE. Approximately 1000 protein spots were detected on the 2DE gels. A representative protein profile is shown in Figure 2. In total, fifteen spots which showed remarkable change (p<0.05) between the model and control groups had been observed, of which 7 spots were upregulated and 8 spots downregulated.

Two-dimensional electrophoresis (2-DE) representative proteomic maps of control
Identification of significantly modified proteins
The proteins extracted from the 2-DE gel were applied to MALDI-TOF/TOF-MS and analyzed by the Mascot search engine to differentially expressed proteins. Finally, we identified 15 proteins which are. shown in Supplementary Table S1 (Supplementary data are available online at www.liebertpub.com/omi). A representative peptide mass fingerprint (PMF) map and database query result of protein prothrombin and transthyretin are shown in Figure 3. Specifically, relative expression of the significantly modified proteins is shown in Figure 4. They appear to be involved in energy production, immunity, metabolism, signal transduction, and chaperoning. Hepatoprotective effects of scoparone action on liver injury was associated with regulated expression of six proteins including Ig kappa chain C, haptoglobin, alpha-1-antitrypsin, zinc finger protein 407, transthyretin, and prothrombin.
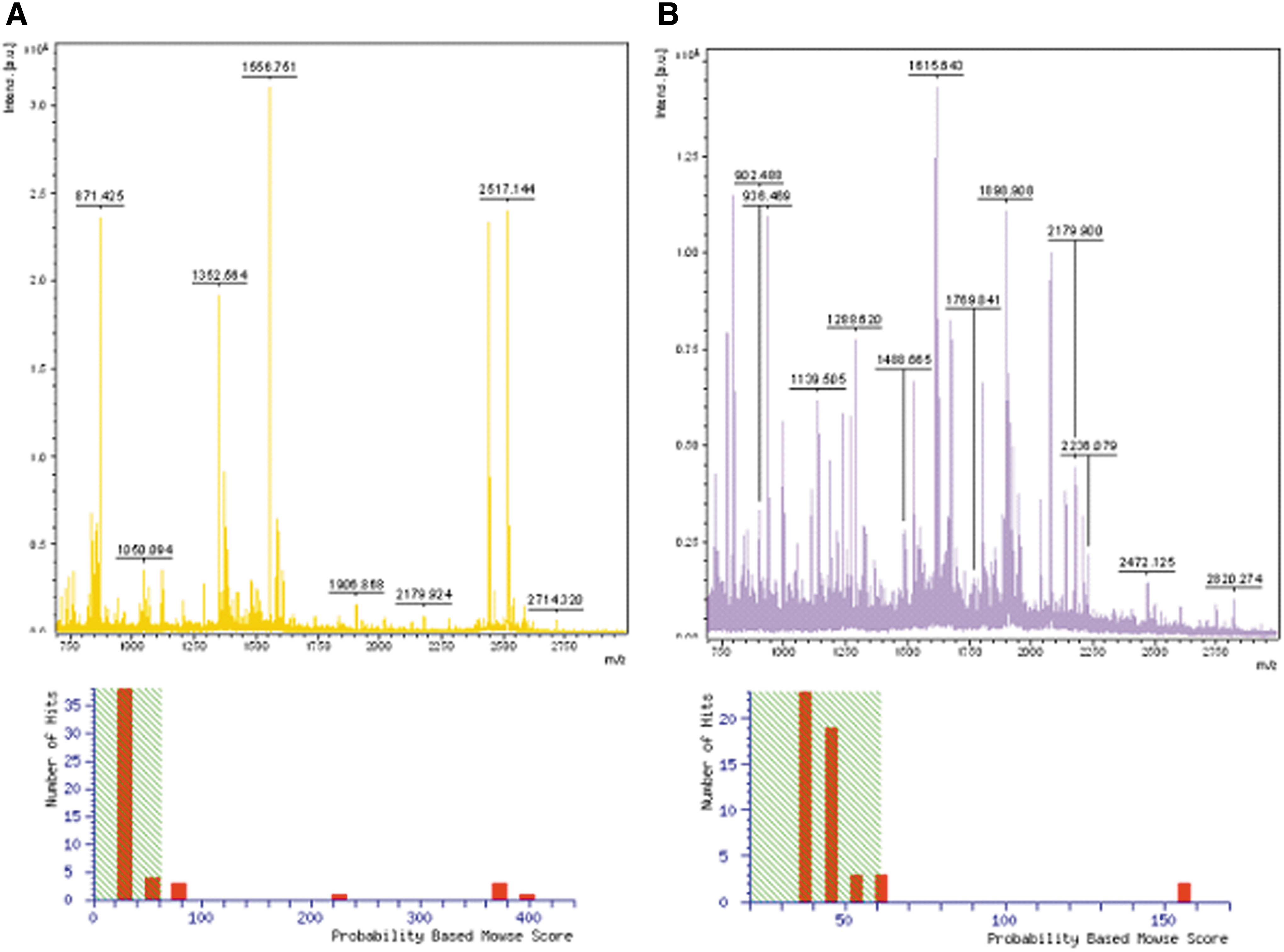
Peptide mass fingerprint spectrum of transthyretin

Relative expression level of the differentially expressed proteins obtained by MALDI-TOF/TOF MS.
Visualization and analysis of protein–protein interaction network
To facilitate access to the protein–protein interaction data, STRING integrates information about interactions from metabolic pathways and drug–target relationships. As seen in Figure 5, association network of six differentially expressed proteins using STRING was constructed, where the nodes are the entities and the edges their interactions. Modes of action are shown in different colors. Differentially expressed proteins are represented as pill-shaped nodes, while proteins are shown as spheres. A possible network associated with scoparone target-related proteins was constructed, suggesting that these proteins were closely related in the protein–protein interaction network. From all the GO categories covered, the identified proteins could be divided into several main groups according to their functions. These closely connected proteins in networks are regarded as the signatures of the underlying targets.

Protein interaction network generated and visualized with STRING 9.0 for proteins identified. Modes of action are shown in different colors. Differentially expressed proteins are represented as pill-shaped nodes, while other proteins are shown as spheres. Nodes that are associated to each other are linked by an edge. Thicker lines represent stronger associations. Lines and for directed edges, arrows of different colors stand for different edge types in the actions view: binding (blue), activation (green), inhibition (red), catalysis (magenta), same activity (cyan), and reaction (black).
Discussion
The tide of drug discovery programs has been turned back to Nature in the search for new drug candidates. Natural products, on the other hand, have evolved to interact with biomolecules, which have been dominating the pharmaceutical industry for a long time, increasingly turning out to be successful. They have contributed to the development of important therapeutics to combat diseases over the past decades (Yang et al., 2012). The value of natural products to drug discovery is still considerable, however, the protein target(s) of action of these fascinating compounds are largely unknown. Recently, MS-based proteomics has the potential to identify novel proteins that are related to the action mechanisms of natural products (Barnett et al., 2012; Schrattenholz et al., 2010; Zhang et al., 2013). Various types of biological networks are being constructed, including protein–protein interaction networks, and protein regulatory networks, to help drug discovery (Abu-Asab et al., 2011; Ge et al., 2012).
Scoparone is an important ingredient for the therapeutic effect of Yinchenhao. Evidence shows that scoparone could yield potential benefits in the treatment and prevention for liver diseases, such as a potent inhibitor of hepatic fibrosis induced by carbon tetrachloride (Wang et al., 2011; Yin et al., 2012; Zhang et al., 2011 and 2012). All these results suggest that it may be a potent drug candidate for further study and up to now, its therapeutic mechanism has still been completely unclear. Therefore, in this study, we have conducted a comprehensive analysis of the possible target proteins following 2-DE and MALDI-TOF/MS. Protein interaction networks were also mapped using STRING analysis program. As a result, hepatoprotective effects of scoparone action against acute hepatic injury rats was associated with regulated expression of six proteins, including Ig kappa chain C, zinc finger protein 407, prothrombin, haptoglobin, alpha-1-antitrypsin, and transthyretin. Furthermore, the change in the levels of these proteins caused by scoparone treatment appear to be involved in antioxidation and signal transduction, energy production, immunity, metabolism, and chaperoning. As seen in Figure 5, association networks of differentially expressed proteins in the scoparone-treated group using STRING was constructed, which may help to increase the understanding of molecular networks for scoparone treatment.
Modern drug discovery is time-consuming and expensive (Savino et al., 2012). The rapid advancement of proteomics approaches has provided a vast of significant information to drug discovery that are recruited for application to pharmaceutical research (Shen et al., 2007). Furthermore, the networks may also play an important role in discovering biomarkers. It has been shown that identified markers are more reproducible if their identification is based on networks rather than single proteins (Heo et al., 2011). Novel protein targets and the protein-associated subsystems provide new insights into the mechanisms underlying pathogenesis. Here, we developed a practical solution particularly to address the bottleneck in the identification of authentic target proteins among a number of candidates. These identifications, undoubtedly, have significantly contributed to the elucidation of the underlying biological mechanism of drug action.
Conclusion
Target-based drug discovery has become a major pharmacological approach since the emergence of -omics technologies. Proteomics holds the key to revolutionize the target identification for natural products. Scoparone is a natural compound with recognized effects against hepatic damage properties. Our study implemented proteomics to identify potential protein targets for liver injury rats affected by scoparone. Six proteins with statistically significant altered expression levels changed under scoparone treatment were identified and might be considered as possible targets of scoparone. These results provide new details on the therapeutic mechanism of scoparone from a molecular perspective. On the other hand, it is unlikely that a compound has only one target protein, and therefore the present method may be useful for the identification of mutli-target proteins that are at the basis of therapeutic effects to the compound. Network-based drug discovery protocols that emphasize the value of small compounds will likely be reevaluated. The methodology reported herein is anticipated to make an important contribution for elucidating the underlying biological mechanism of action at a molecular level toward discovery research in this area.
Footnotes
Acknowledgments
This work was supported by grants from the Key Program of Natural Science Foundation of State (Grant No. 90709019), National Specific Program on the Subject of Public Welfare (Grant No. 200807014), National Key Subject of Drug Innovation (Grant No. 2009ZX09502-005), and National Program on Key Basic Research Project of China (Grant No. 2005CB523406).
Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.